

Effective elimination of fludarabine-resistant CLL cells by PEITC through a redox-mediated mechanism
- PMID: 18574029
- PMCID: PMC2518893
- DOI: 10.1182/blood-2008-04-149815
Effective elimination of fludarabine-resistant CLL cells by PEITC through a redox-mediated mechanism
Abstract
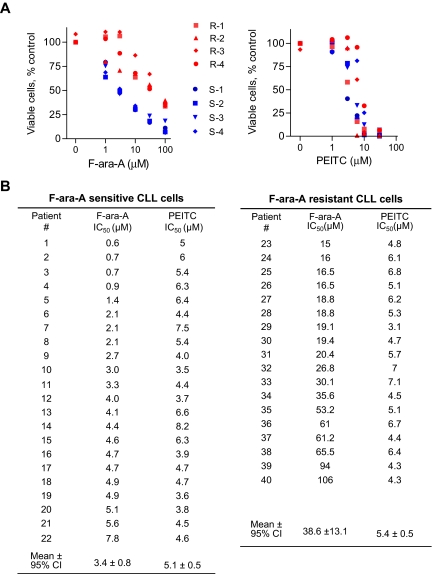
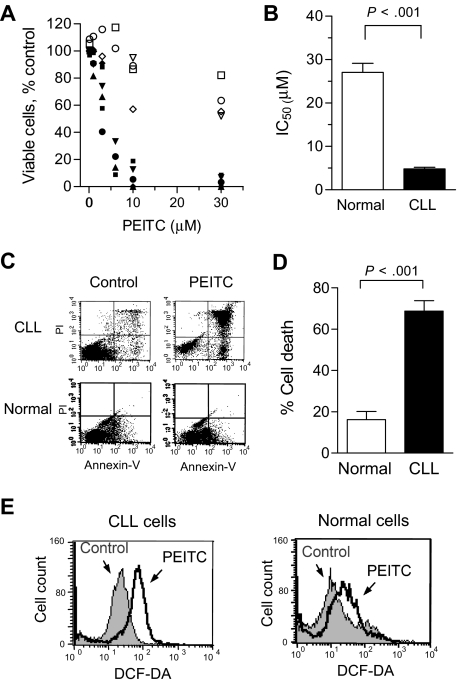
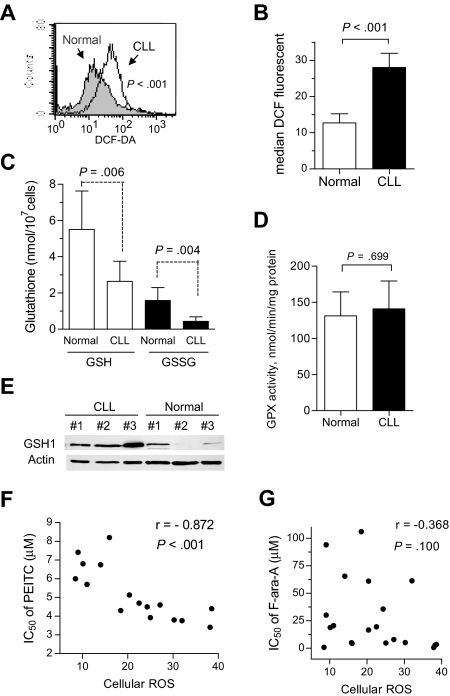
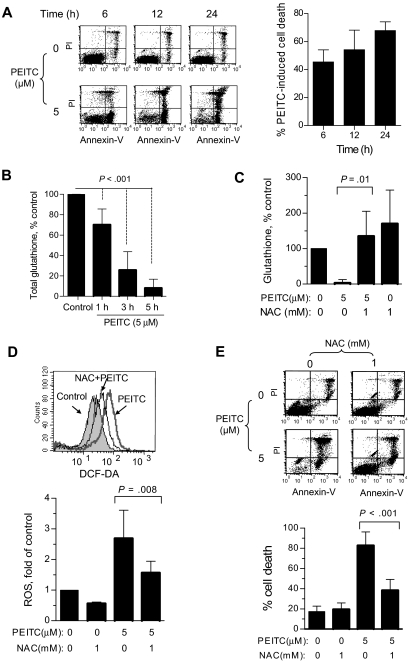
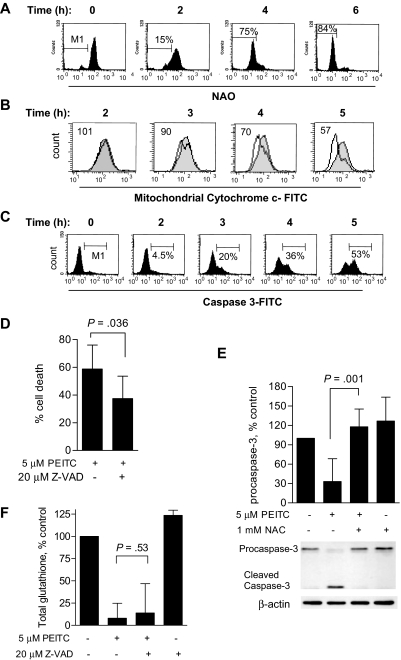
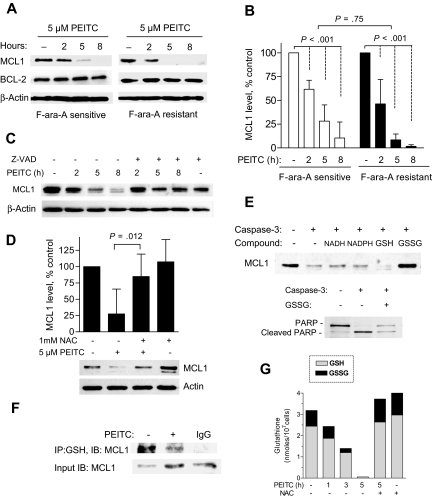
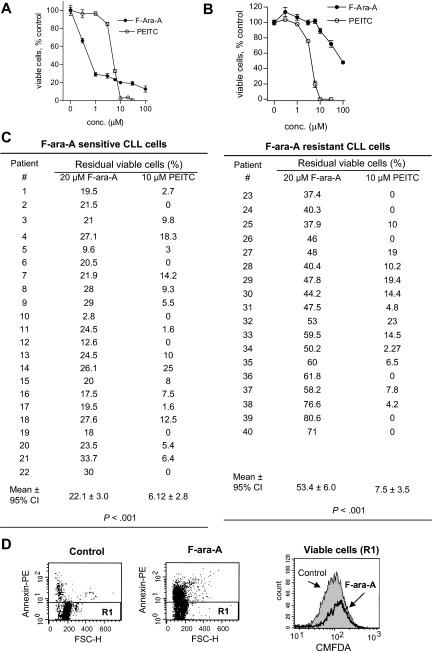
Chronic lymphocytic leukemia (CLL) is the most common adult leukemia, and resistance to fludarabine-based therapies is a major challenge in CLL treatment. Because CLL cells are known to have elevated levels of reactive oxygen species (ROS), we aimed to test a novel ROS-mediated strategy to eliminate fludarabine-resistant CLL cells based on this redox alteration. Using primary CLL cells and normal lymphocytes from patients (n = 58) and healthy subjects (n = 12), we showed that both fludarabine-resistant and -sensitive CLL cells were highly sensitive to beta-phenylethyl isothiocyanate (PEITC) with mean IC(50) values of 5.4 microM and 5.1 microM, respectively. Normal lymphocytes were significantly less sensitive to PEITC (IC(50) = 27 microM, P < .001). CLL cells exhibited intrinsically higher ROS level and lower cellular glutathione, which were shown to be the critical determinants of CLL sensitivity to PEITC. Exposure of CLL cells to PEITC induced severe glutathione depletion, ROS accumulation, and oxidation of mitochondrial cardiolipin leading to massive cell death. Such ROS stress also caused deglutathionylation of MCL1, followed by a rapid degradation of this cell survival molecule. Our study demonstrated that the natural compound PEITC is effective in eliminating fludarabine-resistant CLL cells through a redox-mediated mechanism with low toxicity to normal lymphocytes, and warrants further clinical evaluation.
Figures
Similar articles
-
Effective elimination of chronic lymphocytic leukemia cells in the stromal microenvironment by a novel drug combination strategy using redox-mediated mechanisms.Mol Med Rep. 2015 Nov;12(5):7374-88. doi: 10.3892/mmr.2015.4364. Epub 2015 Sep 25. Mol Med Rep. 2015. PMID: 26458979 Free PMC article.
-
Targeting p53-deficient chronic lymphocytic leukemia cells in vitro and in vivo by ROS-mediated mechanism.Oncotarget. 2016 Nov 1;7(44):71378-71389. doi: 10.18632/oncotarget.12110. Oncotarget. 2016. PMID: 27655686 Free PMC article.
-
Effective killing of Gleevec-resistant CML cells with T315I mutation by a natural compound PEITC through redox-mediated mechanism.Leukemia. 2008 Jun;22(6):1191-9. doi: 10.1038/leu.2008.74. Epub 2008 Apr 3. Leukemia. 2008. PMID: 18385754 Free PMC article.
-
Metabolic reprogramming in the CLL TME; potential for new therapeutic targets.Semin Hematol. 2024 Jun;61(3):155-162. doi: 10.1053/j.seminhematol.2024.02.001. Epub 2024 Feb 15. Semin Hematol. 2024. PMID: 38493076 Review.
-
Protein Phosphorylation and Redox Status: An as Yet Elusive Dyad in Chronic Lymphocytic Leukemia.Cancers (Basel). 2022 Oct 6;14(19):4881. doi: 10.3390/cancers14194881. Cancers (Basel). 2022. PMID: 36230804 Free PMC article. Review.
Cited by
-
Loss of p53 and altered miR15-a/16-1MCL-1 pathway in CLL: insights from TCL1-Tg:p53(-/-) mouse model and primary human leukemia cells.Leukemia. 2014 Jan;28(1):118-28. doi: 10.1038/leu.2013.125. Epub 2013 Apr 23. Leukemia. 2014. PMID: 23608884 Free PMC article.
-
Systems biology modeling reveals a possible mechanism of the tumor cell death upon oncogene inactivation in EGFR addicted cancers.PLoS One. 2011;6(12):e28930. doi: 10.1371/journal.pone.0028930. Epub 2011 Dec 14. PLoS One. 2011. PMID: 22194952 Free PMC article.
-
N',N'-Dimethyl-N',N'-bis(phenylcarbonothioyl) Propanedihydrazide (Elesclomol) Selectively Kills Cisplatin Resistant Lung Cancer Cells through Reactive Oxygen Species (ROS).Cancers (Basel). 2009;1(1):23-38. doi: 10.3390/cancers1010023. Cancers (Basel). 2009. PMID: 20535236 Free PMC article.
-
Expression of TOMM34 and Its Clinicopathological Correlations in Urothelial Carcinoma of the Bladder.Pathol Oncol Res. 2020 Jan;26(1):411-418. doi: 10.1007/s12253-018-0524-3. Epub 2018 Oct 31. Pathol Oncol Res. 2020. PMID: 30382527
-
Targeting Energy Metabolism in Cancer Treatment.Int J Mol Sci. 2022 May 16;23(10):5572. doi: 10.3390/ijms23105572. Int J Mol Sci. 2022. PMID: 35628385 Free PMC article. Review.
References
-
- Keating MJ, Chiorazzi N, Messmer B, et al. Biology and treatment of chronic lymphocytic leukemia. Hematology Am Soc Hematol Educ Program. 2003;1:153–175. - PubMed
-
- Elter T, Hallek M, Engert A. Fludarabine in chronic lymphocytic leukaemia. Expert Opin Pharmacother. 2006;7:1641–1651. - PubMed
-
- Byrd JC, Lin TS, Grever MR. Treatment of relapsed chronic lymphocytic leukemia: old and new therapies. Semin Oncol. 2006;33:210–219. - PubMed
-
- Turgut B, Vural O, Pala FS, et al. 17p Deletion is associated with resistance of B-cell chronic lymphocytic leukemia cells to in vitro fludarabine-induced apoptosis. Leuk Lymphoma. 2007;48:311–220. - PubMed
-
- Grever MR, Lucas DM, Dewald GW, et al. Comprehensive assessment of genetic and molecular features predicting outcome in patients with chronic lymphocytic leukemia: results from the US Intergroup Phase III Trial E2997. J Clin Oncol. 2007;25:799–804. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
