

Critical involvement of the ATM-dependent DNA damage response in the apoptotic demise of HIV-1-elicited syncytia
- PMID: 18560558
- PMCID: PMC2423469
- DOI: 10.1371/journal.pone.0002458
Critical involvement of the ATM-dependent DNA damage response in the apoptotic demise of HIV-1-elicited syncytia
Abstract
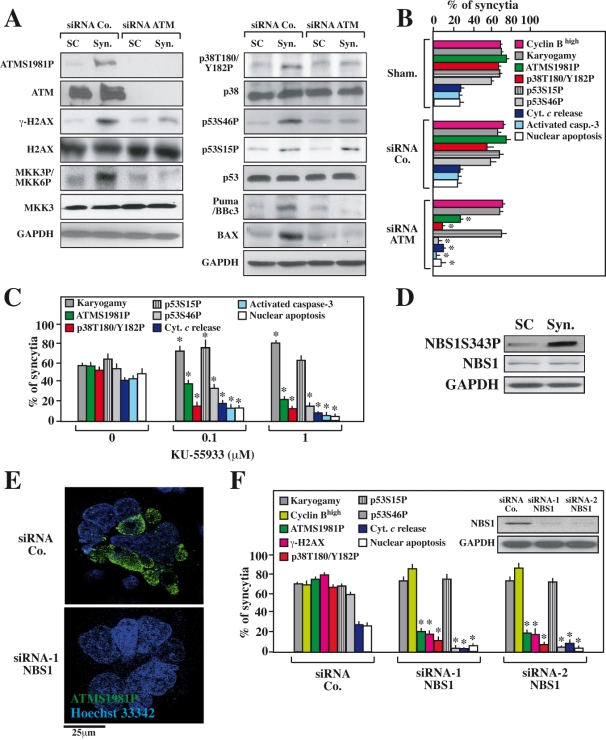
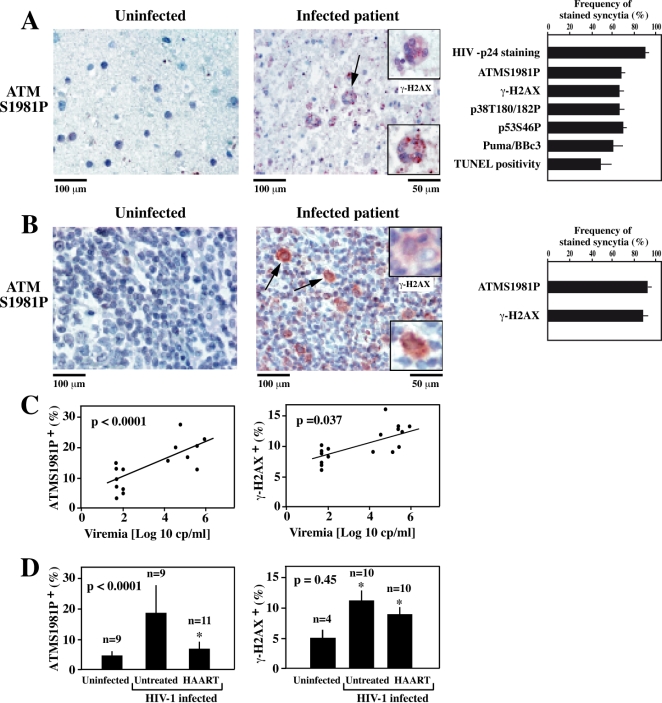
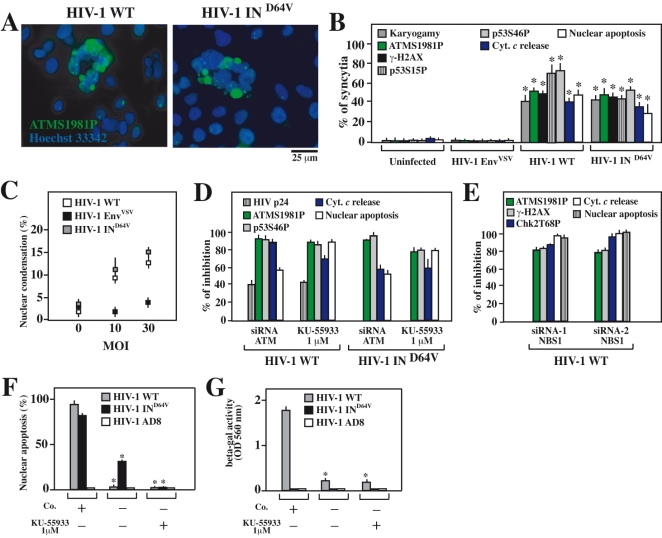
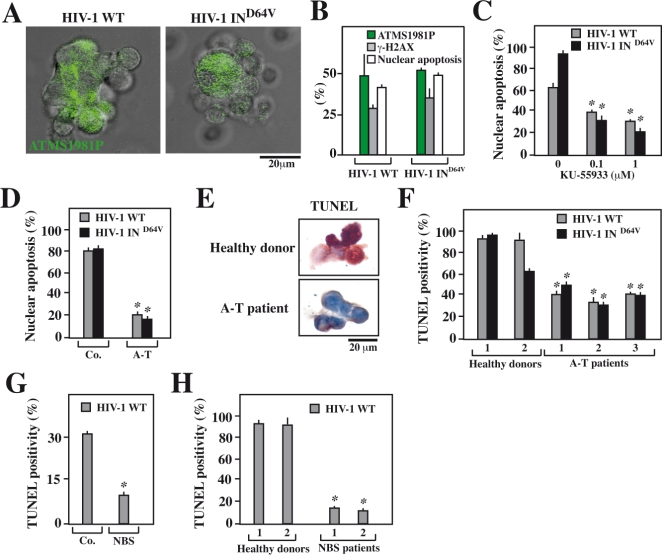
DNA damage can activate the oncosuppressor protein ataxia telangiectasia mutated (ATM), which phosphorylates the histone H2AX within characteristic DNA damage foci. Here, we show that ATM undergoes an activating phosphorylation in syncytia elicited by the envelope glycoprotein complex (Env) of human immunodeficiency virus-1 (HIV-1) in vitro. This was accompanied by aggregation of ATM in discrete nuclear foci that also contained phospho-histone H2AX. DNA damage foci containing phosphorylated ATM and H2AX were detectable in syncytia present in the brain or lymph nodes from patients with HIV-1 infection, as well as in a fraction of blood leukocytes, correlating with viral status. Knockdown of ATM or of its obligate activating factor NBS1 (Nijmegen breakage syndrome 1 protein), as well as pharmacological inhibition of ATM with KU-55933, inhibited H2AX phosphorylation and prevented Env-elicited syncytia from undergoing apoptosis. ATM was found indispensable for the activation of MAP kinase p38, which catalyzes the activating phosphorylation of p53 on serine 46, thereby causing p53 dependent apoptosis. Both wild type HIV-1 and an HIV-1 mutant lacking integrase activity induced syncytial apoptosis, which could be suppressed by inhibiting ATM. HIV-1-infected T lymphoblasts from patients with inactivating ATM or NBS1 mutations also exhibited reduced syncytial apoptosis. Altogether these results indicate that apoptosis induced by a fusogenic HIV-1 Env follows a pro-apoptotic pathway involving the sequential activation of ATM, p38MAPK and p53.
Conflict of interest statement
Figures



Similar articles
-
Pro-apoptotic function of checkpoint kinase-2 in syncytia elicited by the HIV-1 envelope.Cell Cycle. 2009 Feb 1;8(3):438-42. doi: 10.4161/cc.8.3.7642. Epub 2009 Feb 15. Cell Cycle. 2009. PMID: 19177012
-
The tumor suppressor protein PML controls apoptosis induced by the HIV-1 envelope.Cell Death Differ. 2009 Feb;16(2):298-311. doi: 10.1038/cdd.2008.158. Epub 2008 Nov 21. Cell Death Differ. 2009. PMID: 19023333
-
53BP1 represses mitotic catastrophe in syncytia elicited by the HIV-1 envelope.Cell Death Differ. 2010 May;17(5):811-20. doi: 10.1038/cdd.2009.159. Epub 2009 Oct 30. Cell Death Differ. 2010. PMID: 19876065
-
Ataxia-telangiectasia-like disorder (ATLD)-its clinical presentation and molecular basis.DNA Repair (Amst). 2004 Aug-Sep;3(8-9):1219-25. doi: 10.1016/j.dnarep.2004.04.009. DNA Repair (Amst). 2004. PMID: 15279810 Review.
-
Cytometry of ATM activation and histone H2AX phosphorylation to estimate extent of DNA damage induced by exogenous agents.Cytometry A. 2007 Sep;71(9):648-61. doi: 10.1002/cyto.a.20426. Cytometry A. 2007. PMID: 17622968 Free PMC article. Review.
Cited by
-
T-Cell Signaling in HIV-1 Infection.Open Virol J. 2013 Jul 26;7:57-71. doi: 10.2174/1874357920130621001. eCollection 2013. Open Virol J. 2013. PMID: 23986795 Free PMC article.
-
The DNA damage response induced by infection with human cytomegalovirus and other viruses.Viruses. 2014 May 23;6(5):2155-85. doi: 10.3390/v6052155. Viruses. 2014. PMID: 24859341 Free PMC article. Review.
-
HIV-1 Tat potently stabilises Mdm2 and enhances viral replication.Biochem J. 2017 Jul 11;474(14):2449-2464. doi: 10.1042/BCJ20160825. Biochem J. 2017. PMID: 28468838 Free PMC article.
-
Syncytial apoptosis signaling network induced by the HIV-1 envelope glycoprotein complex: an overview.Cell Death Dis. 2015 Aug 6;6(8):e1846. doi: 10.1038/cddis.2015.204. Cell Death Dis. 2015. PMID: 26247731 Free PMC article. Review.
-
Viral manipulation of DNA repair and cell cycle checkpoints.DNA Repair (Amst). 2009 Sep 2;8(9):1166-76. doi: 10.1016/j.dnarep.2009.04.016. Epub 2009 May 26. DNA Repair (Amst). 2009. PMID: 19473887 Free PMC article. Review.
References
-
- Hamers FF, Downs AM. The changing face of the HIV epidemic in western Europe: what are the implications for public health policies? Lancet. 2004;364:83–94. - PubMed
-
- Wyen C, Hoffmann C, Schmeisser N, Wohrmann A, Qurishi N, et al. Progressive multifocal leukencephalopathy in patients on highly active antiretroviral therapy: survival and risk factors of death. J Acquir Immune Defic Syndr. 2004;37:1263–1268. - PubMed
-
- Cook JE, Dasgupta S, Middaugh LD, Terry EC, Gorry PR, et al. Highly active antiretroviral therapy and human immunodeficiency virus encephalitis. Ann Neurol. 2005;57:795–803. - PubMed
-
- Badley AD, Pilon AA, Landay A, Lynch DH. Mechanisms of HIV-associated lymphocyte apoptosis. Blood. 2000;96:2951–2964. - PubMed
-
- Pantaleo G, Fauci AS. Apoptosis in HIV infection. Nat Med. 1995;1:118–120. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
