

Regulation of autophagy by cytoplasmic p53
- PMID: 18454141
- PMCID: PMC2676564
- DOI: 10.1038/ncb1730
Regulation of autophagy by cytoplasmic p53
Abstract
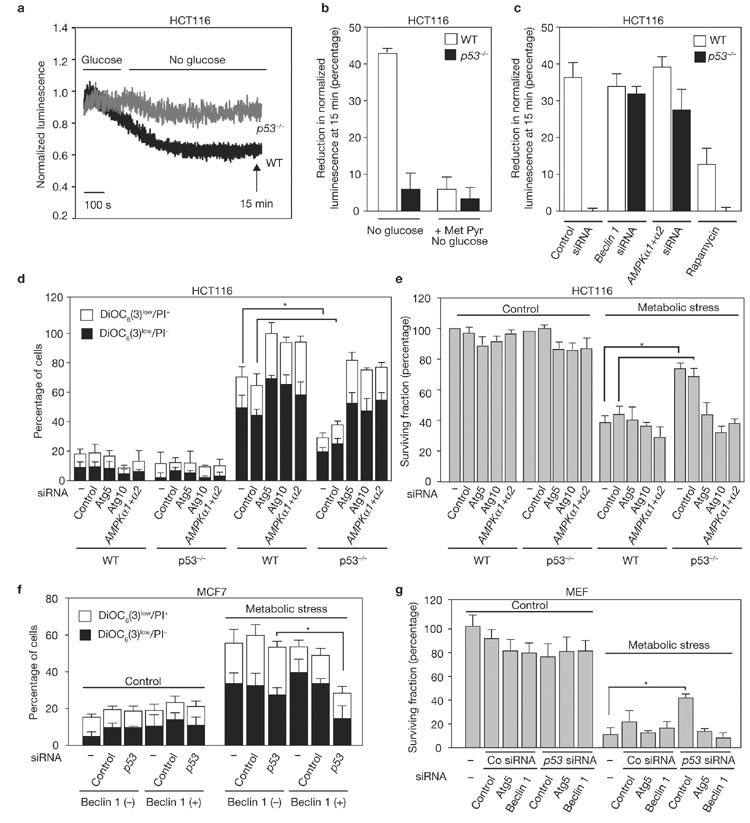
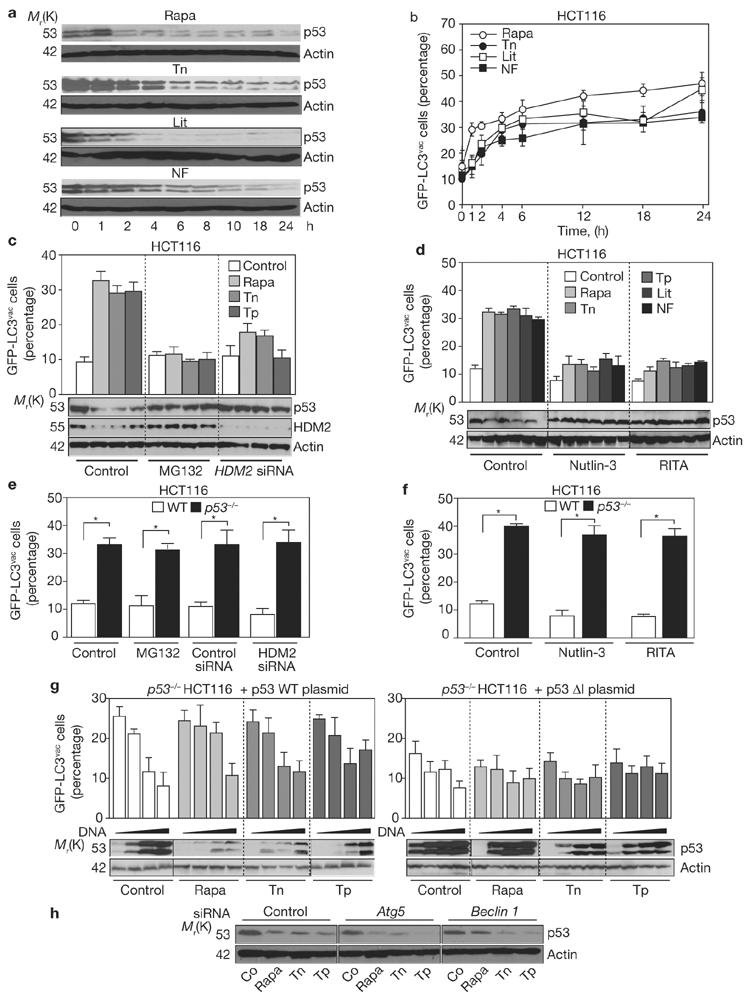
Multiple cellular stressors, including activation of the tumour suppressor p53, can stimulate autophagy. Here we show that deletion, depletion or inhibition of p53 can induce autophagy in human, mouse and nematode cells subjected to knockout, knockdown or pharmacological inhibition of p53. Enhanced autophagy improved the survival of p53-deficient cancer cells under conditions of hypoxia and nutrient depletion, allowing them to maintain high ATP levels. Inhibition of p53 led to autophagy in enucleated cells, and cytoplasmic, not nuclear, p53 was able to repress the enhanced autophagy of p53(-/-) cells. Many different inducers of autophagy (for example, starvation, rapamycin and toxins affecting the endoplasmic reticulum) stimulated proteasome-mediated degradation of p53 through a pathway relying on the E3 ubiquitin ligase HDM2. Inhibition of p53 degradation prevented the activation of autophagy in several cell lines, in response to several distinct stimuli. These results provide evidence of a key signalling pathway that links autophagy to the cancer-associated dysregulation of p53.
Figures





Similar articles
-
A dual role of p53 in the control of autophagy.Autophagy. 2008 Aug;4(6):810-4. doi: 10.4161/auto.6486. Epub 2008 Jun 24. Autophagy. 2008. PMID: 18604159
-
Cytoplasmic destruction of p53 by the endoplasmic reticulum-resident ubiquitin ligase 'Synoviolin'.EMBO J. 2007 Jan 10;26(1):113-22. doi: 10.1038/sj.emboj.7601490. Epub 2006 Dec 14. EMBO J. 2007. PMID: 17170702 Free PMC article.
-
Nuclear and cytoplasmic degradation of endogenous p53 and HDM2 occurs during down-regulation of the p53 response after multiple types of DNA damage.FASEB J. 2003 Sep;17(12):1622-30. doi: 10.1096/fj.02-0931com. FASEB J. 2003. PMID: 12958168
-
p53 proteasomal degradation: poly-ubiquitination is not the whole story.Cell Cycle. 2005 Aug;4(8):1015-8. doi: 10.4161/cc.4.8.1900. Epub 2005 Aug 7. Cell Cycle. 2005. PMID: 16082197 Review.
-
p53 signaling and autophagy in cancer: a revolutionary strategy could be developed for cancer treatment.Autophagy. 2011 Jun;7(6):565-71. doi: 10.4161/auto.7.6.14073. Epub 2011 Jun 1. Autophagy. 2011. PMID: 21099252 Review.
Cited by
-
PML at Mitochondria-Associated Membranes Is Critical for the Repression of Autophagy and Cancer Development.Cell Rep. 2016 Aug 30;16(9):2415-27. doi: 10.1016/j.celrep.2016.07.082. Epub 2016 Aug 18. Cell Rep. 2016. PMID: 27545895 Free PMC article.
-
The Effects of Methylene Blue on Autophagy and Apoptosis in MRI-Defined Normal Tissue, Ischemic Penumbra and Ischemic Core.PLoS One. 2015 Jun 29;10(6):e0131929. doi: 10.1371/journal.pone.0131929. eCollection 2015. PLoS One. 2015. PMID: 26121129 Free PMC article.
-
Signaling and other functions of lipids in autophagy: a review.Lipids Health Dis. 2020 Sep 30;19(1):214. doi: 10.1186/s12944-020-01389-2. Lipids Health Dis. 2020. PMID: 32998777 Free PMC article. Review.
-
Potential relationship between Sirt3 and autophagy in ovarian cancer.Oncol Lett. 2020 Nov;20(5):162. doi: 10.3892/ol.2020.12023. Epub 2020 Aug 26. Oncol Lett. 2020. PMID: 32934730 Free PMC article. Review.
-
MDM2 and MDMX: Alone and together in regulation of p53.Transl Cancer Res. 2012 Aug;1(2):88-89. Transl Cancer Res. 2012. PMID: 23002429 Free PMC article.
References
-
- Qu X, et al. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. 2007;128:931–946. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
