

Distinct roles for p107 and p130 in Rb-independent cellular senescence
- PMID: 18418057
- PMCID: PMC3474322
- DOI: 10.4161/cc.7.9.5945
Distinct roles for p107 and p130 in Rb-independent cellular senescence
Abstract
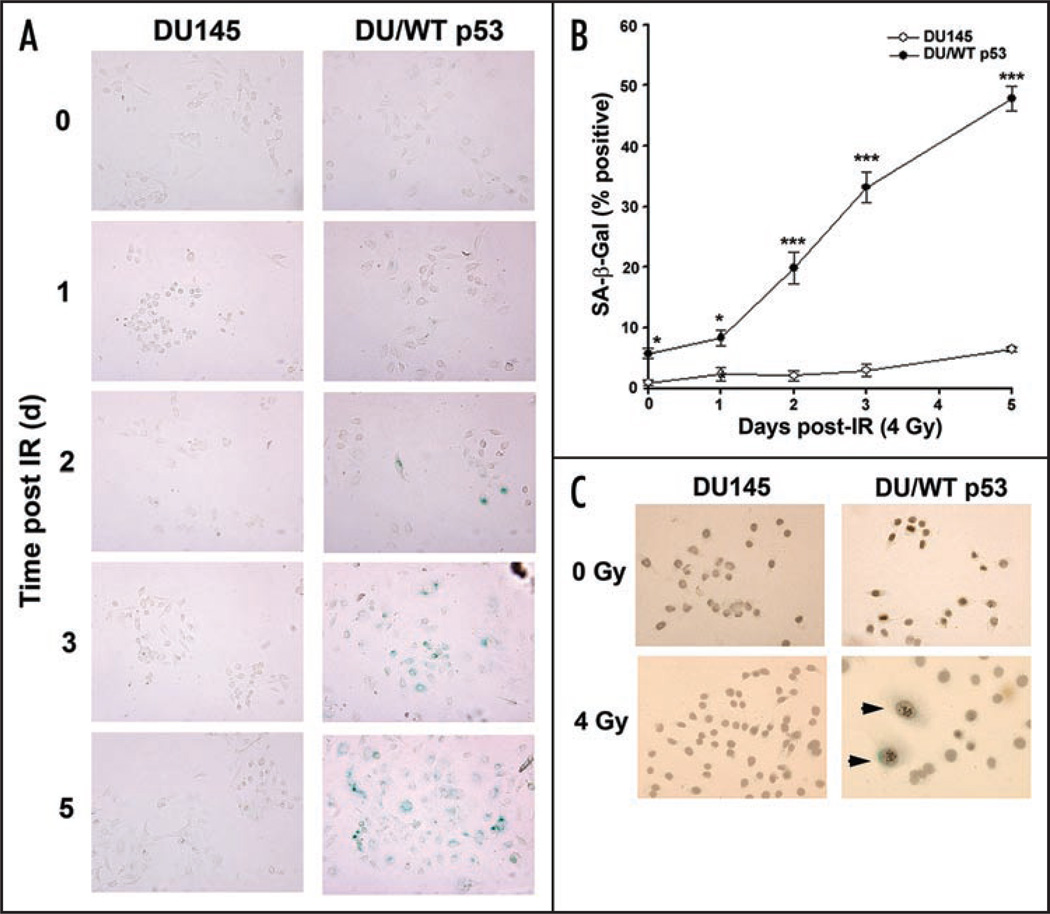
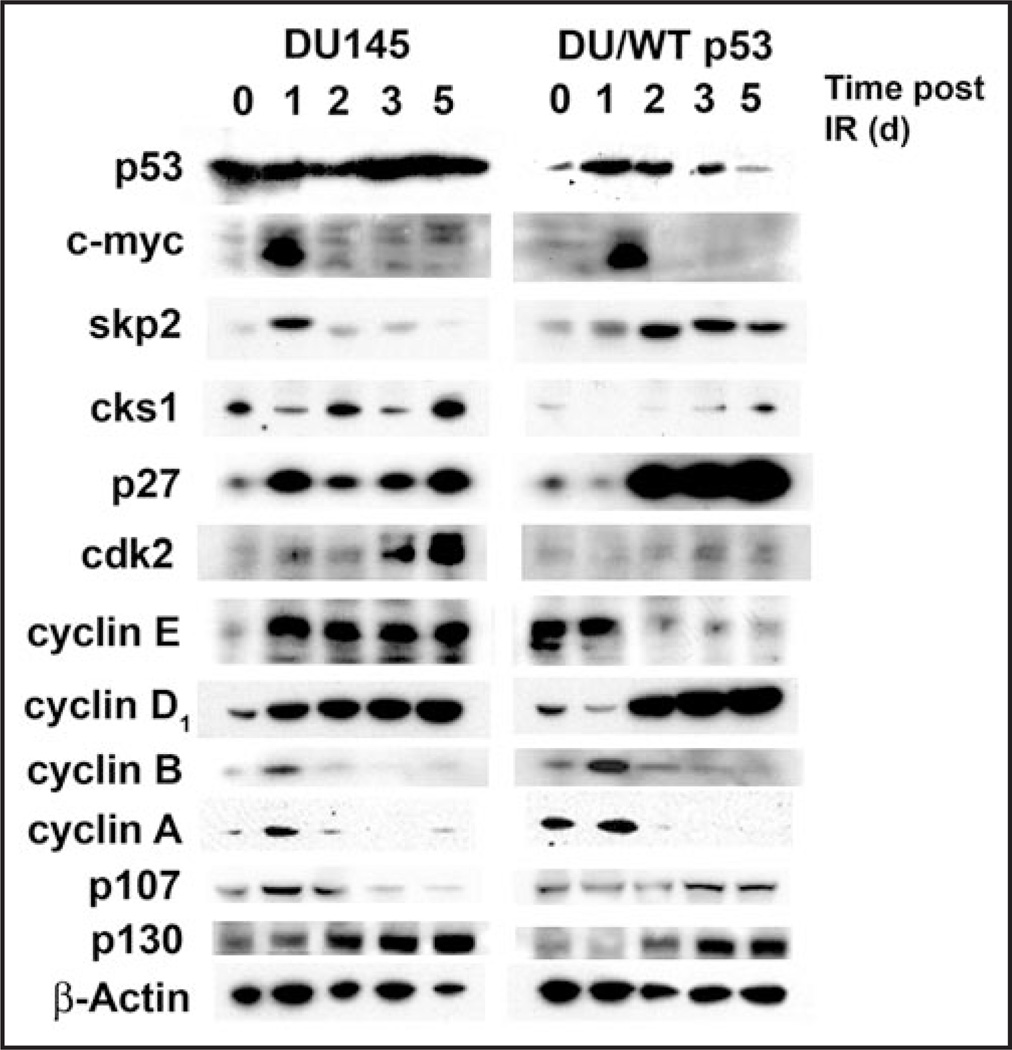
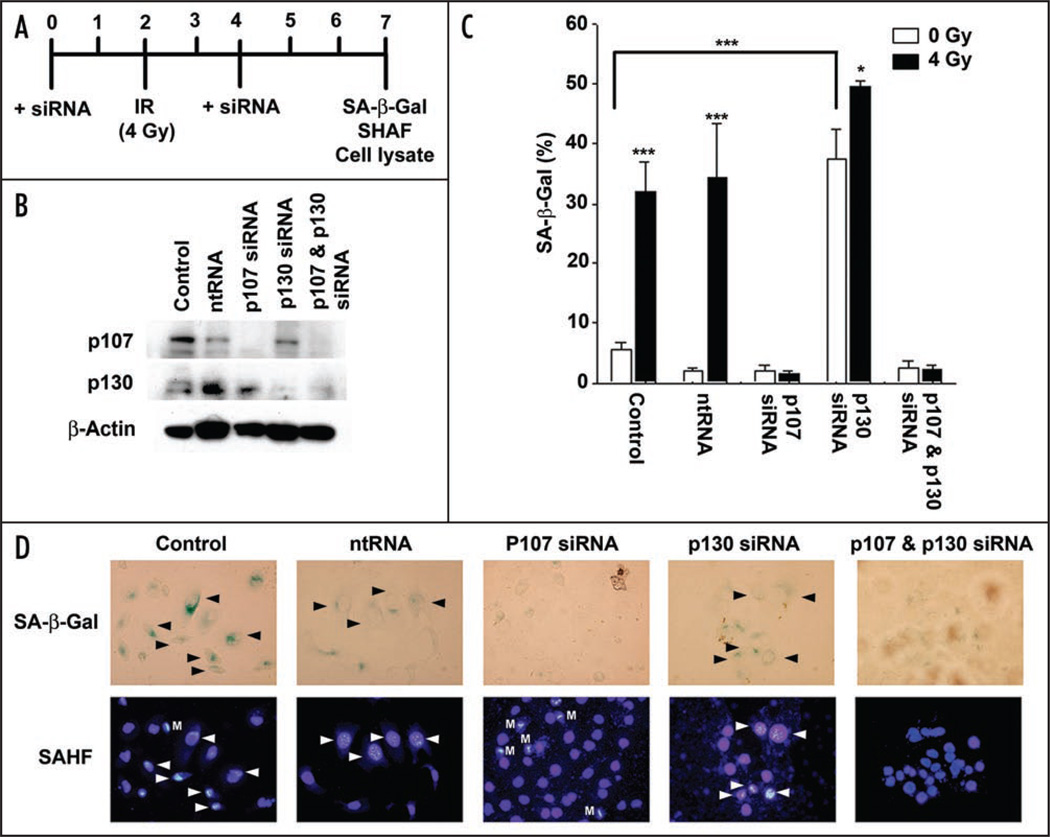
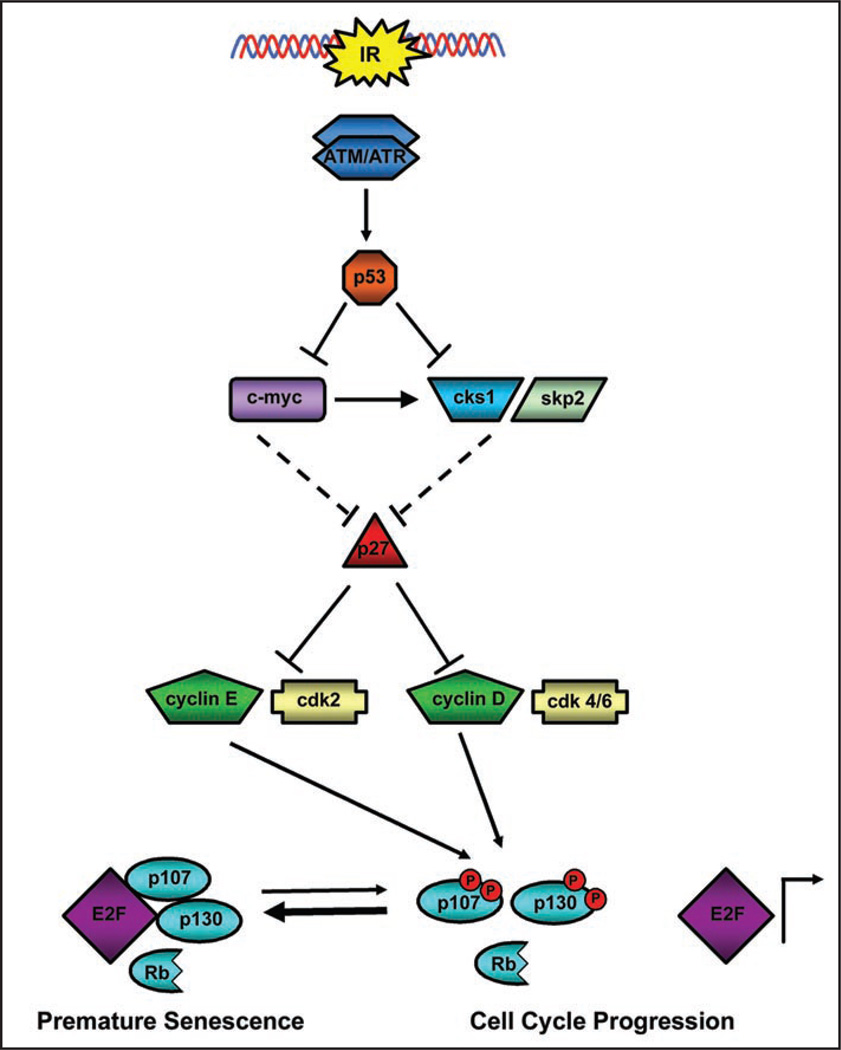
Telomere attrition, DNA damage and constitutive mitogenic signaling can all trigger cellular senescence in normal cells and serve as a defense against tumor progression. Cancer cells may circumvent this cellular defense by acquiring genetic mutations in checkpoint proteins responsible for regulating permanent cell cycle arrest. A small family of tumor suppressor genes encoding the retinoblastoma susceptibility protein family (Rb, p107, p130) exerts a partially redundant control of entry into S phase of DNA replication and cellular proliferation. Here we report that activation of the p53-dependent DNA damage response has been found to accelerate senescence in human prostate cancer cells lacking a functional Rb protein. This novel form of irradiation-induced premature cellular senescence reinforces the notion that other Rb family members may compensate for loss of Rb protein in the DNA damage response pathway. Consistent with this hypothesis, depletion of p107 potently inhibits the irradiation-induced senescence observed in DU145 cells. In contrast, p130 depletion triggers a robust and unexpected form of premature senescence in unirradiated cells. The dominant effect of depleting both p107 and p130, in the absence of Rb, was a complete blockade of irradiation-induced cellular senescence. Onset of the p107-dependent senescence was temporally associated with p53-mediated stabilization of the cyclin-dependent kinase inhibitor p27 and decreases in c-myc and cks1 expression. These results indicate that p107 is required for initiation of accelerated cellular senescence in the absence of Rb and introduces the concept that p130 may be required to prevent the onset of terminal growth arrest in unstimulated prostate cancer cells lacking a functional Rb allele.
Figures
Similar articles
-
RB, p130 and p107 differentially repress G1/S and G2/M genes after p53 activation.Nucleic Acids Res. 2019 Dec 2;47(21):11197-11208. doi: 10.1093/nar/gkz961. Nucleic Acids Res. 2019. PMID: 31667499 Free PMC article.
-
Retinoblastoma gene-independent G1 phase arrest by flavone, phosphatidylinositol 3-kinase inhibitor, and histone deacetylase inhibitor.Cancer Sci. 2012 Dec;103(12):2139-43. doi: 10.1111/cas.12012. Epub 2012 Oct 26. Cancer Sci. 2012. PMID: 22957647 Free PMC article.
-
The specific role of pRb in p16 (INK4A) -mediated arrest of normal and malignant human breast cells.Cell Cycle. 2012 Mar 1;11(5):1008-13. doi: 10.4161/cc.11.5.19492. Epub 2012 Mar 1. Cell Cycle. 2012. PMID: 22333593 Free PMC article.
-
Regulation of the retinoblastoma-E2F pathway by the ubiquitin-proteasome system.Biochim Biophys Acta. 2015 Oct;1849(10):1289-97. doi: 10.1016/j.bbagrm.2015.08.008. Epub 2015 Aug 28. Biochim Biophys Acta. 2015. PMID: 26319102 Review.
-
The Rb family connects with the Tp53 family in skin carcinogenesis.Mol Carcinog. 2007 Aug;46(8):618-23. doi: 10.1002/mc.20338. Mol Carcinog. 2007. PMID: 17486638 Review.
Cited by
-
The cell fate: senescence or quiescence.Mol Biol Rep. 2016 Nov;43(11):1213-1220. doi: 10.1007/s11033-016-4065-0. Epub 2016 Aug 24. Mol Biol Rep. 2016. PMID: 27558094 Review.
-
The relative contributions of the p53 and pRb pathways in oncogene-induced melanocyte senescence.Aging (Albany NY). 2009 May 16;1(6):542-56. doi: 10.18632/aging.100051. Aging (Albany NY). 2009. PMID: 20157537 Free PMC article.
-
Silencing of protein kinase D2 induces glioma cell senescence via p53-dependent and -independent pathways.Neuro Oncol. 2014 Jul;16(7):933-45. doi: 10.1093/neuonc/not303. Neuro Oncol. 2014. PMID: 24463355 Free PMC article.
-
Role of senescence and mitotic catastrophe in cancer therapy.Cell Div. 2010 Jan 21;5:4. doi: 10.1186/1747-1028-5-4. Cell Div. 2010. Retraction in: Cell Div. 2012 Jul 12;7:15. doi: 10.1186/1747-1028-7-15. PMID: 20205872 Free PMC article. Retracted.
-
What's so special about RB?Cancer Cell. 2010 Apr 13;17(4):313-4. doi: 10.1016/j.ccr.2010.03.010. Cancer Cell. 2010. PMID: 20385355 Free PMC article.
References
-
- Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007 - PubMed
-
- Lehmann BD, McCubrey JA, Jefferson HS, Paine MS, Chappell WH, Terrian DM. A dominant role for p53-dependent cellular senescence in radiosensitization of human prostate cancer cells. Cell Cycle. 2007;6:595–605. - PubMed
-
- Di Leonardo A, Linke SP, Clarkin K, Wahl GM. DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev. 1994;8:2540–2551. - PubMed
-
- Narita M, Nunez S, Heard E, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous