

Idiopathic pulmonary fibrosis
- PMID: 18366757
- PMCID: PMC2330030
- DOI: 10.1186/1750-1172-3-8
Idiopathic pulmonary fibrosis
Abstract
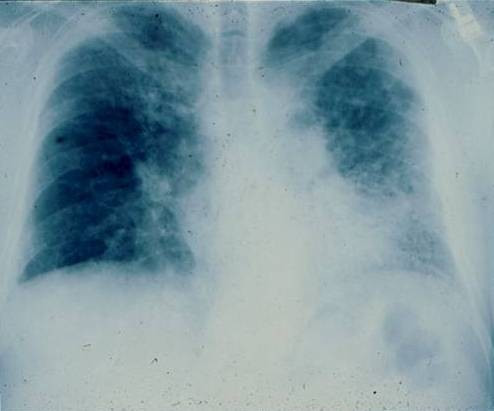
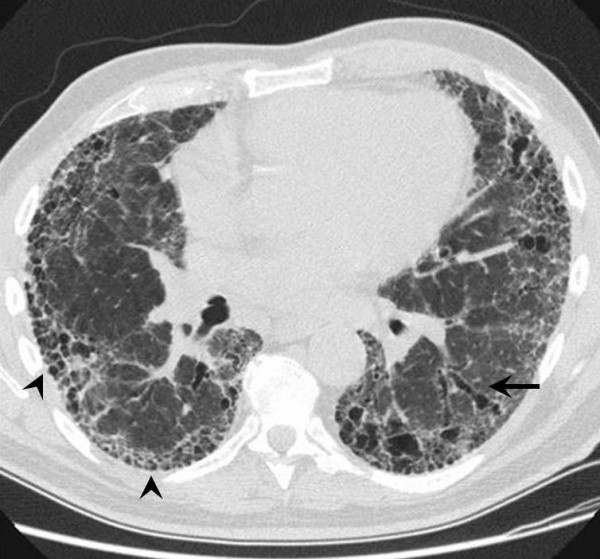
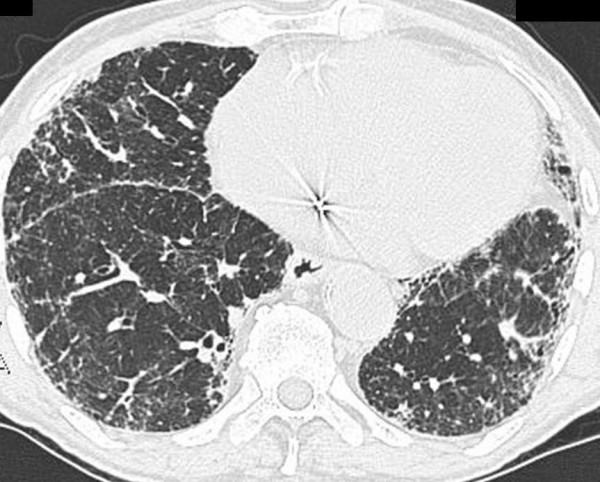
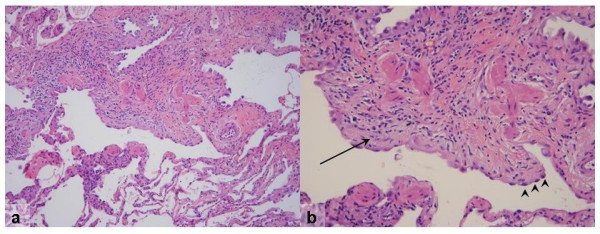
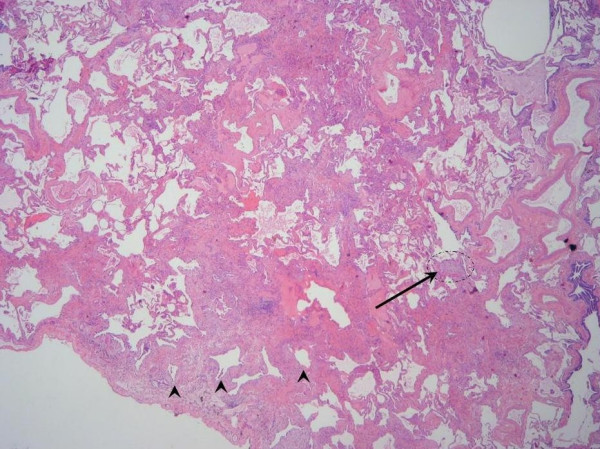
Idiopathic pulmonary fibrosis (IPF) is a non-neoplastic pulmonary disease that is characterized by the formation of scar tissue within the lungs in the absence of any known provocation. IPF is a rare disease which affects approximately 5 million persons worldwide. The prevalence is estimated to be slightly greater in men (20.2/100,000) than in women (13.2/100,000). The mean age at presentation is 66 years. IPF initially manifests with symptoms of exercise-induced breathless and dry coughing. Auscultation of the lungs reveals early inspiratory crackles, predominantly located in the lower posterior lung zones upon physical exam. Clubbing is found in approximately 50% of IPF patients. Cor pulmonale develops in association with end-stage disease. In that case, classic signs of right heart failure may be present. Etiology remains incompletely understood. Some environmental factors may be associated with IPF (cigarette smoking, exposure to silica and livestock). IPF is recognized on high-resolution computed tomography by peripheral, subpleural lower lobe reticular opacities in association with subpleural honeycomb changes. IPF is associated with a pathological lesion known as usual interstitial pneumonia (UIP). The UIP pattern consists of normal lung alternating with patches of dense fibrosis, taking the form of collagen sheets. The diagnosis of IPF requires correlation of the clinical setting with radiographic images and a lung biopsy. In the absence of lung biopsy, the diagnosis of IPF can be made by defined clinical criteria that were published in guidelines endorsed by several professional societies. Differential diagnosis includes other idiopathic interstitial pneumonia, connective tissue diseases (systemic sclerosis, polymyositis, rheumatoid arthritis), forme fruste of autoimmune disorders, chronic hypersensitivity pneumonitis and other environmental (sometimes occupational) exposures. IPF is typically progressive and leads to significant disability. The median survival is 2 to 5 years from the time of diagnosis. Medical therapy is ineffective in the treatment of IPF. New molecular therapeutic targets have been identified and several clinical trials are investigating the efficacy of novel medication. Meanwhile, pulmonary transplantation remains a viable option for patients with IPF. It is expected that, during the next decade, considerable progress will be made toward the understanding and treatment of this devastating illness.
Figures
Similar articles
-
Usual interstitial pneumonia end-stage features from explants with radiologic and pathological correlations.Ann Diagn Pathol. 2015 Aug;19(4):269-76. doi: 10.1016/j.anndiagpath.2015.05.003. Epub 2015 May 13. Ann Diagn Pathol. 2015. PMID: 26025258
-
Diagnosis and Management of Idiopathic Pulmonary Fibrosis.R I Med J (2013). 2021 Sep 1;104(7):26-29. R I Med J (2013). 2021. PMID: 34437662 Review.
-
Clinical impact of the radiological indeterminate for usual interstitial pneumonia pattern on the diagnosis of idiopathic pulmonary fibrosis.Respir Investig. 2021 Jan;59(1):81-89. doi: 10.1016/j.resinv.2020.07.001. Epub 2020 Aug 28. Respir Investig. 2021. PMID: 32868263
-
Clinical features of usual interstitial pneumonia with anti-neutrophil cytoplasmic antibody in comparison with idiopathic pulmonary fibrosis.Respirology. 2016 Jul;21(5):920-6. doi: 10.1111/resp.12763. Epub 2016 Mar 19. Respirology. 2016. PMID: 26994375
-
Pulmonary fibrosis.Methods Mol Med. 2005;117:3-44. doi: 10.1385/1-59259-940-0:003. Methods Mol Med. 2005. PMID: 16130230 Free PMC article. Review.
Cited by
-
B7H3-dependent myeloid-derived suppressor cell recruitment and activation in pulmonary fibrosis.Front Immunol. 2022 Aug 15;13:901349. doi: 10.3389/fimmu.2022.901349. eCollection 2022. Front Immunol. 2022. PMID: 36045668 Free PMC article.
-
A fully automated image analysis method to quantify lung fibrosis in the bleomycin-induced rat model.PLoS One. 2018 Mar 16;13(3):e0193057. doi: 10.1371/journal.pone.0193057. eCollection 2018. PLoS One. 2018. PMID: 29547661 Free PMC article.
-
Idiopathic Pulmonary Fibrosis: Pathogenesis and the Emerging Role of Long Non-Coding RNAs.Int J Mol Sci. 2020 Jan 14;21(2):524. doi: 10.3390/ijms21020524. Int J Mol Sci. 2020. PMID: 31947693 Free PMC article. Review.
-
Buyang Huanwu Decoction Ameliorates Bleomycin-Induced Pulmonary Fibrosis in Rats via Downregulation of Related Protein and Gene Expression.Evid Based Complement Alternat Med. 2018 Feb 28;2018:9185485. doi: 10.1155/2018/9185485. eCollection 2018. Evid Based Complement Alternat Med. 2018. PMID: 29681987 Free PMC article.
-
Design of a Study Assessing Disease Behaviour During the Peri-Diagnostic Period in Patients with Interstitial Lung Disease: The STARLINER Study.Adv Ther. 2019 Jan;36(1):232-243. doi: 10.1007/s12325-018-0845-3. Epub 2018 Nov 30. Adv Ther. 2019. PMID: 30506309 Free PMC article.
References
-
- American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS) Am J Respir Crit Care Med. 2000;161:646–664. - PubMed
-
- Katzenstein AL, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med. 1998;157:1301–1315. - PubMed
-
- Flaherty KR, Mumford JA, Murray S, Kazerooni EA, Gross BH, Colby TV, Travis WD, Flint A, Toews GB, Lynch JP, 3rd, Martinez FJ. Prognostic implications of physiologic and radiographic changes in idiopathic interstitial pneumonia. Am J Respir Crit Care Med. 2003;168:543–548. doi: 10.1164/rccm.200209-1112OC. - DOI - PubMed
-
- Latsi PI, du Bois RM, Nicholson AG, Colby TV, Bisirtzoglou D, Nikolakopoulou A, Veeraraghavan S, Hansell DM, Wells AU. Fibrotic idiopathic interstitial pneumonia: the prognostic value of longitudinal functional trends. Am J Respir Crit Care Med. 2003;168:531–537. doi: 10.1164/rccm.200210-1245OC. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
