

Velvet: algorithms for de novo short read assembly using de Bruijn graphs
- PMID: 18349386
- PMCID: PMC2336801
- DOI: 10.1101/gr.074492.107
Velvet: algorithms for de novo short read assembly using de Bruijn graphs
Abstract
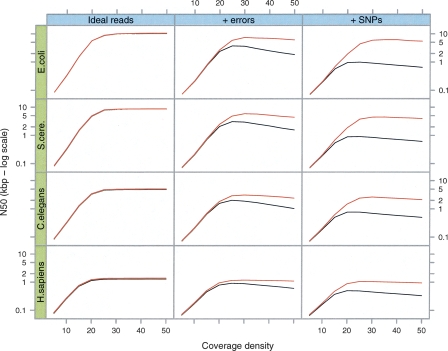
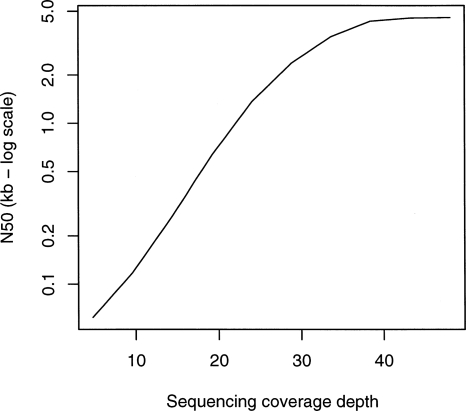
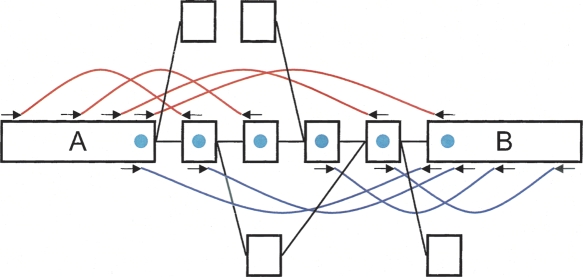
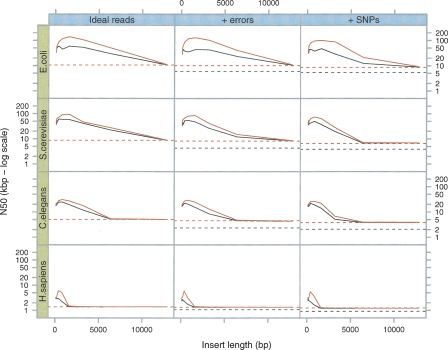
We have developed a new set of algorithms, collectively called "Velvet," to manipulate de Bruijn graphs for genomic sequence assembly. A de Bruijn graph is a compact representation based on short words (k-mers) that is ideal for high coverage, very short read (25-50 bp) data sets. Applying Velvet to very short reads and paired-ends information only, one can produce contigs of significant length, up to 50-kb N50 length in simulations of prokaryotic data and 3-kb N50 on simulated mammalian BACs. When applied to real Solexa data sets without read pairs, Velvet generated contigs of approximately 8 kb in a prokaryote and 2 kb in a mammalian BAC, in close agreement with our simulated results without read-pair information. Velvet represents a new approach to assembly that can leverage very short reads in combination with read pairs to produce useful assemblies.
Figures
Similar articles
-
Benchmarking of de novo assembly algorithms for Nanopore data reveals optimal performance of OLC approaches.BMC Genomics. 2016 Aug 22;17 Suppl 7(Suppl 7):507. doi: 10.1186/s12864-016-2895-8. BMC Genomics. 2016. PMID: 27556636 Free PMC article.
-
Efficient parallel and out of core algorithms for constructing large bi-directed de Bruijn graphs.BMC Bioinformatics. 2010 Nov 15;11:560. doi: 10.1186/1471-2105-11-560. BMC Bioinformatics. 2010. PMID: 21078174 Free PMC article.
-
ALLPATHS: de novo assembly of whole-genome shotgun microreads.Genome Res. 2008 May;18(5):810-20. doi: 10.1101/gr.7337908. Epub 2008 Mar 13. Genome Res. 2008. PMID: 18340039 Free PMC article.
-
The present and future of de novo whole-genome assembly.Brief Bioinform. 2018 Jan 1;19(1):23-40. doi: 10.1093/bib/bbw096. Brief Bioinform. 2018. PMID: 27742661 Review.
-
De novo assembly of short sequence reads.Brief Bioinform. 2010 Sep;11(5):457-72. doi: 10.1093/bib/bbq020. Epub 2010 Aug 19. Brief Bioinform. 2010. PMID: 20724458 Review.
Cited by
-
Plastid phylogenomics of Robinsonia (Senecioneae; Asteraceae), endemic to the Juan Fernández Islands: insights into structural organization and molecular evolution.BMC Plant Biol. 2024 Oct 28;24(1):1016. doi: 10.1186/s12870-024-05711-3. BMC Plant Biol. 2024. PMID: 39465373 Free PMC article.
-
New persistent plant RNA virus carries mutations to weaken viral suppression of antiviral RNA interference.Mol Plant Pathol. 2024 Oct;25(10):e70020. doi: 10.1111/mpp.70020. Mol Plant Pathol. 2024. PMID: 39462907 Free PMC article.
-
Genetic variation, structural analysis, and virulence implications of BimA and BimC in clinical isolates of Burkholderia pseudomallei in Thailand.Sci Rep. 2024 Oct 23;14(1):24966. doi: 10.1038/s41598-024-74922-3. Sci Rep. 2024. PMID: 39443499 Free PMC article.
-
A novel sequence type of carbapenem-resistant hypervirulent Klebsiella pneumoniae strains from a county-level tertiary hospital in Southeastern China.Medicine (Baltimore). 2024 Oct 18;103(42):e40120. doi: 10.1097/MD.0000000000040120. Medicine (Baltimore). 2024. PMID: 39432660 Free PMC article.
References
-
- Batzoglou S. Algorithmic challenges in mammalian genome sequence assembly. In: Dunn M., et al., editors. Encyclopedia of genomics, proteomics and bioinformatics. John Wiley and Sons; New York: 2005. Part 4.
-
- Batzoglou S., Jaffe D.B., Stanley K., Butler J., Gnerre S., Mauceli E., Berger B., Mesirov J.P., Lander E.S., Jaffe D.B., Stanley K., Butler J., Gnerre S., Mauceli E., Berger B., Mesirov J.P., Lander E.S., Stanley K., Butler J., Gnerre S., Mauceli E., Berger B., Mesirov J.P., Lander E.S., Butler J., Gnerre S., Mauceli E., Berger B., Mesirov J.P., Lander E.S., Gnerre S., Mauceli E., Berger B., Mesirov J.P., Lander E.S., Mauceli E., Berger B., Mesirov J.P., Lander E.S., Berger B., Mesirov J.P., Lander E.S., Mesirov J.P., Lander E.S., Lander E.S. ARACHNE: A whole genome shotgun assembler. Genome Res. 2002;12:177–189. - PMC - PubMed
-
- Bentley D.R. Whole-genome re-sequencing. Curr. Opin. Genet. Dev. 2006;16:545–552. - PubMed
-
- Bokhari S.H., Sauer J.R., Sauer J.R. A parallel graph decomposition algorithm for DNA sequencing with nanopores. Bioinformatics. 2005;21:889–896. - PubMed
-
- Chaisson M., Pevzner P.A., Tang H., Pevzner P.A., Tang H., Tang H. Fragment assembly with short reads. Bioinformatics. 2004;20:2067–2074. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources