

Activation of nicotinic acetylcholine receptor prevents the production of reactive oxygen species in fibrillar beta amyloid peptide (1-42)-stimulated microglia
- PMID: 18305393
- PMCID: PMC2679317
- DOI: 10.3858/emm.2008.40.1.11
Activation of nicotinic acetylcholine receptor prevents the production of reactive oxygen species in fibrillar beta amyloid peptide (1-42)-stimulated microglia
Abstract
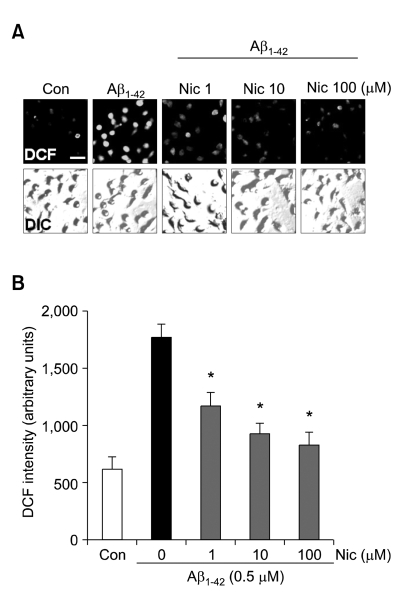
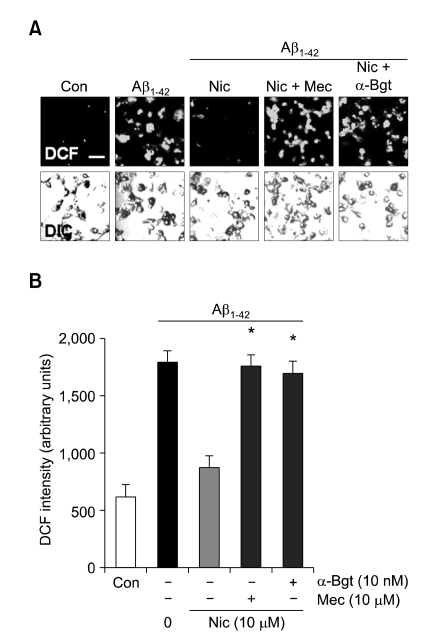
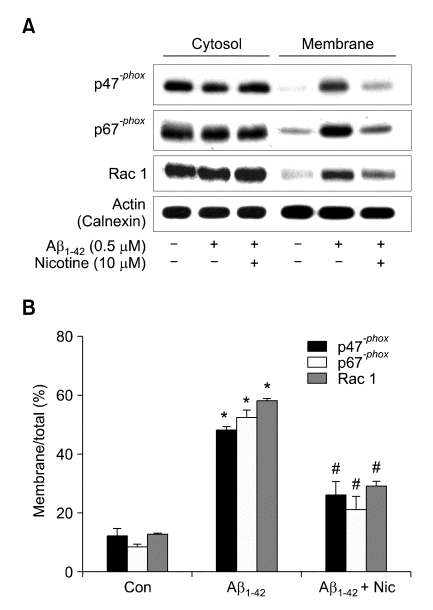
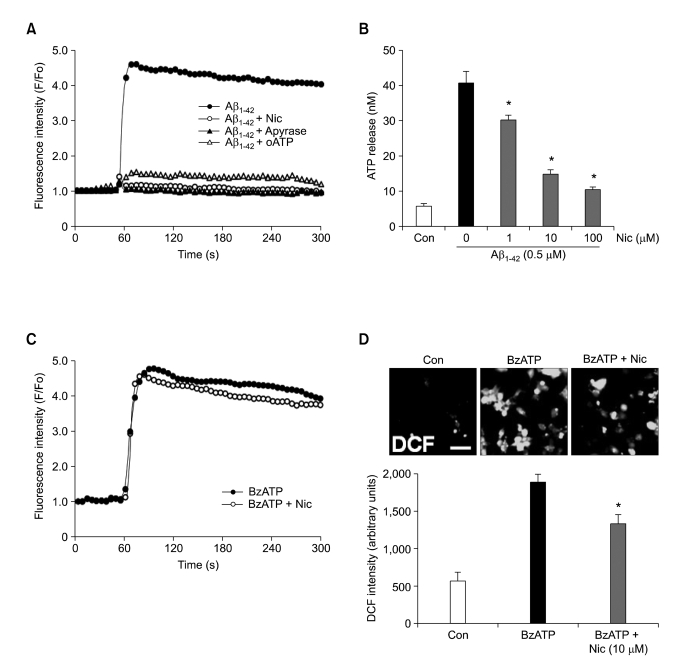
Recent studies have reported that the cholinergic anti-inflammatory pathway regulates peripheral inflammatory responses via alpha7 nicotinic acetylcholine receptors (alpha7 nAChRs) and that acetylcholine and nicotine regulate the expression of proinflammatory mediators such as TNF-alpha and prostaglandin E2 in microglial cultures. In a previous study we showed that ATP released by beta-amyloid-stimulated microglia induced reactive oxygen species (ROS) production, in a process involving the P2X(7) receptor (P2X(7)R), in an autocrine fashion. These observations led us to investigate whether stimulation by nicotine could regulate fibrillar beta amyloid peptide (1-42) (fAbeta1-42)-induced ROS production by modulating ATP efflux-mediated Ca(2+) influx through P2X(7)R. Nicotine inhibited ROS generation in fAbeta(1-42)-stimulated microglial cells, and this inhibition was blocked by mecamylamine, a non-selective nAChR antagonist, and a-bungarotoxin, a selective alpha7 nAChR antagonist. Nicotine inhibited NADPH oxidase activation and completely blocked Ca(2+) influx in fAbeta(1-42)-stimulated microglia. Moreover, ATP release from fAbeta(1-42)-stimulated microglia was significantly suppressed by nicotine treatment. In contrast, nicotine did not inhibit 2',3'-O-(4-benzoyl)-benzoyl ATP (BzATP)-induced Ca(2+) influx, but inhibited ROS generation in BzATP-stimulated microglia, indicating an inhibitory effect of nicotine on a signaling process downstream of P2X(7)R. Taken together, these results suggest that the inhibitory effect of nicotine on ROS production in fAbeta1-42-stimulated microglia is mediated by indirect blockage of ATP release and by directly altering the signaling process downstream from P2X(7)R.
Figures
Similar articles
-
ATP released from beta-amyloid-stimulated microglia induces reactive oxygen species production in an autocrine fashion.Exp Mol Med. 2007 Dec 31;39(6):820-7. doi: 10.1038/emm.2007.89. Exp Mol Med. 2007. PMID: 18160853
-
Microglial alpha7 nicotinic acetylcholine receptors drive a phospholipase C/IP3 pathway and modulate the cell activation toward a neuroprotective role.J Neurosci Res. 2006 Jun;83(8):1461-70. doi: 10.1002/jnr.20850. J Neurosci Res. 2006. PMID: 16652343
-
P2X(7) receptors exert a permissive role on the activation of release-enhancing presynaptic alpha7 nicotinic receptors co-existing on rat neocortex glutamatergic terminals.Neuropharmacology. 2006 May;50(6):705-13. doi: 10.1016/j.neuropharm.2005.11.016. Epub 2006 Jan 19. Neuropharmacology. 2006. PMID: 16427662
-
Purinergic mediated changes in Ca2+ mobilization and functional responses in microglia: effects of low levels of ATP.J Neurosci Res. 2005 Aug 1;81(3):349-56. doi: 10.1002/jnr.20475. J Neurosci Res. 2005. PMID: 15948175 Review.
-
Nicotinic regulation of microglia: potential contributions to addiction.J Neural Transm (Vienna). 2024 May;131(5):425-435. doi: 10.1007/s00702-023-02703-9. Epub 2023 Oct 1. J Neural Transm (Vienna). 2024. PMID: 37778006 Review.
Cited by
-
Astrocytic and microglial nicotinic acetylcholine receptors: an overlooked issue in Alzheimer's disease.J Neural Transm (Vienna). 2016 Dec;123(12):1359-1367. doi: 10.1007/s00702-016-1580-z. Epub 2016 Jun 4. J Neural Transm (Vienna). 2016. PMID: 27262818 Review.
-
Neurotransmitter receptors on microglia.Stroke Vasc Neurol. 2016 Jun 24;1(2):52-58. doi: 10.1136/svn-2016-000012. eCollection 2016 Jun. Stroke Vasc Neurol. 2016. PMID: 28959464 Free PMC article. Review.
-
A G protein-coupled α7 nicotinic receptor regulates signaling and TNF-α release in microglia.FEBS Open Bio. 2017 Aug 7;7(9):1350-1361. doi: 10.1002/2211-5463.12270. eCollection 2017 Sep. FEBS Open Bio. 2017. PMID: 28904864 Free PMC article.
-
NADPH oxidases in oxidant production by microglia: activating receptors, pharmacology and association with disease.Br J Pharmacol. 2017 Jun;174(12):1733-1749. doi: 10.1111/bph.13425. Epub 2016 Feb 26. Br J Pharmacol. 2017. PMID: 26750203 Free PMC article. Review.
-
Role of vascular risk factors and vascular dysfunction in Alzheimer's disease.Mt Sinai J Med. 2010 Jan-Feb;77(1):82-102. doi: 10.1002/msj.20155. Mt Sinai J Med. 2010. PMID: 20101718 Free PMC article. Review.
References
-
- Ballerini P, Di Iorio P, Ciccarelli R, Nargi E, D'Alimonte I, Traversa U, Rathbone MP, Caciagli F. Glial cells express multiple ATP binding cassette proteins which are involved in ATP release. Neuroreport. 2002;13:1789–1792. - PubMed
-
- Bianca VD, Dusi S, Bianchini E, Dal Pra I, Rossi F. β-amyloid activates the O-2 forming NADPH oxidase in microglia, monocytes, and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer's disease. J Biol Chem. 1999;274:15493–15499. - PubMed
-
- Buisson B, Bertrand D. Nicotine addiction: the possible role of functional upregulation. Trends Pharmacol Sci. 2002;23:130–136. - PubMed
-
- Burghaus L, Schutz U, Krempel U, de Vos RA, Jansen Steur EN, Wevers A, Lindstrom J, Schroder H. Quantitative assessment of nicotinic acetylcholine receptor proteins in the cerebral cortex of Alzheimer patients. Brain Res Mol Brain Res. 2000;76:385–388. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous