

Nucleotide excision repair deficient mouse models and neurological disease
- PMID: 18272436
- PMCID: PMC2474780
- DOI: 10.1016/j.dnarep.2007.12.006
Nucleotide excision repair deficient mouse models and neurological disease
Abstract
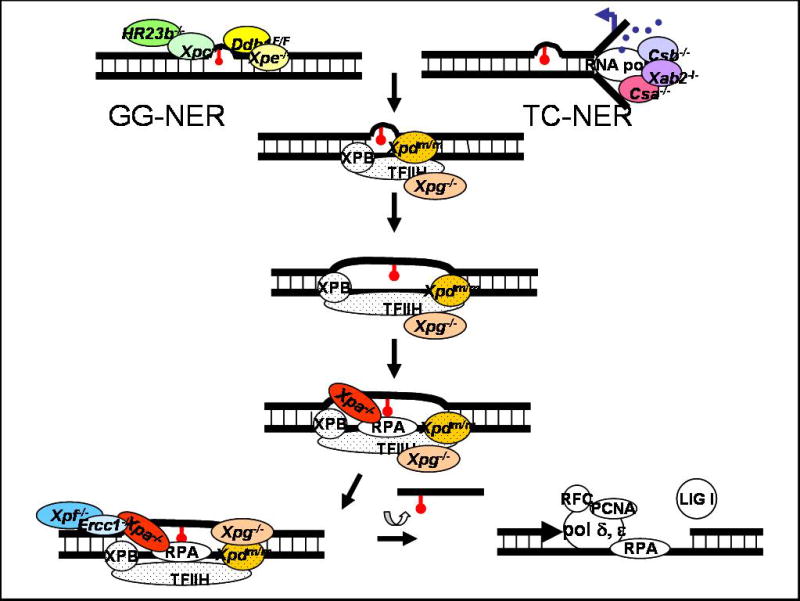
Nucleotide excision repair (NER) is a highly conserved mechanism to remove helix-distorting DNA base damage. A major substrate for NER is DNA damage caused by environmental genotoxins, most notably ultraviolet radiation. Xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy are three human diseases caused by inherited defects in NER. The symptoms and severity of these diseases vary dramatically, ranging from profound developmental delay to cancer predisposition and accelerated aging. All three syndromes include neurological disease, indicating an important role for NER in protecting against spontaneous DNA damage as well. To study the pathophysiology caused by DNA damage, numerous mouse models of NER-deficiency were generated by knocking-out genes required for NER or knocking-in disease-causing human mutations. This review explores the utility of these mouse models to study neurological disease caused by NER-deficiency.
Figures
Similar articles
-
Nucleotide excision repair genes shaping embryonic development.Open Biol. 2019 Oct 31;9(10):190166. doi: 10.1098/rsob.190166. Epub 2019 Oct 30. Open Biol. 2019. PMID: 31662099 Free PMC article. Review.
-
Tissue-specific accelerated aging in nucleotide excision repair deficiency.Mech Ageing Dev. 2008 Jul-Aug;129(7-8):408-15. doi: 10.1016/j.mad.2008.04.010. Epub 2008 May 1. Mech Ageing Dev. 2008. PMID: 18538374 Free PMC article. Review.
-
Age-related neuronal degeneration: complementary roles of nucleotide excision repair and transcription-coupled repair in preventing neuropathology.PLoS Genet. 2011 Dec;7(12):e1002405. doi: 10.1371/journal.pgen.1002405. Epub 2011 Dec 8. PLoS Genet. 2011. PMID: 22174697 Free PMC article.
-
Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype-phenotype relationship.Neuroscience. 2007 Apr 14;145(4):1388-96. doi: 10.1016/j.neuroscience.2006.12.020. Epub 2007 Feb 1. Neuroscience. 2007. PMID: 17276014 Free PMC article. Review.
-
Nucleotide excision repair and neurological diseases.DNA Repair (Amst). 2008 Jul 1;7(7):1155-67. doi: 10.1016/j.dnarep.2008.03.015. Epub 2008 May 5. DNA Repair (Amst). 2008. PMID: 18456575 Review.
Cited by
-
Mutations in Cockayne Syndrome-Associated Genes (Csa and Csb) Predispose to Cisplatin-Induced Hearing Loss in Mice.J Neurosci. 2016 Apr 27;36(17):4758-70. doi: 10.1523/JNEUROSCI.3890-15.2016. J Neurosci. 2016. PMID: 27122034 Free PMC article.
-
Atrx deficiency induces telomere dysfunction, endocrine defects, and reduced life span.J Clin Invest. 2013 May;123(5):2049-63. doi: 10.1172/JCI65634. Epub 2013 Apr 8. J Clin Invest. 2013. PMID: 23563309 Free PMC article.
-
Inefficient DNA Repair Is an Aging-Related Modifier of Parkinson's Disease.Cell Rep. 2016 May 31;15(9):1866-75. doi: 10.1016/j.celrep.2016.04.071. Epub 2016 May 19. Cell Rep. 2016. PMID: 27210754 Free PMC article.
-
Nucleotide Excision Repair in Caenorhabditis elegans.Mol Biol Int. 2011;2011:542795. doi: 10.4061/2011/542795. Epub 2011 Aug 17. Mol Biol Int. 2011. PMID: 22091407 Free PMC article.
-
Defective transcription initiation causes postnatal growth failure in a mouse model of nucleotide excision repair (NER) progeria.Proc Natl Acad Sci U S A. 2012 Feb 21;109(8):2995-3000. doi: 10.1073/pnas.1114941109. Epub 2012 Feb 8. Proc Natl Acad Sci U S A. 2012. PMID: 22323595 Free PMC article.
References
-
- Sugasawa K, Ng JM, Masutani C, Maekawa T, Uchida A, van der Spek PJ, Eker AP, Rademakers S, Visser C, Aboussekhra A, Wood RD, Hanaoka F, Bootsma D, Hoeijmakers JH. Two human homologs of Rad23 are functionally interchangeable in complex formation and stimulation of XPC repair activity. Mol Cell Biol. 1997;17:6924–6931. - PMC - PubMed
-
- Sugasawa K. UV-induced ubiquitylation of XPC complex, the UV-DDB-ubiquitin ligase complex, and DNA repair. J Mol Histol. 2006;37:189–202. - PubMed
-
- Laine JP, Egly JM. When transcription and repair meet: a complex system. Trends Genet. 2006;22:430–436. - PubMed
-
- Ito S, Kuraoka I, Chymkowitch P, Compe E, Takedachi A, Ishigami C, Coin F, Egly JM, Tanaka K. XPG stabilizes TFIIH, allowing transactivation of nuclear receptors: implications for Cockayne syndrome in XP-G/CS patients. Mol Cell. 2007;26:231–243. - PubMed
-
- Giglia-Mari G, Coin F, Ranish JA, Hoogstraten D, Theil A, Wijgers N, Jaspers NG, Raams A, Argentini M, van der Spek PJ, Botta E, Stefanini M, Egly JM, Aebersold R, Hoeijmakers JHJ, Vermeulen W. A new, tenth subunit of TFIIH is responsible for the DNA repair syndrome trichothiodystrophy group A. Nat Genet. 2004;36:714–719. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
