

Pancreatic cancer
- PMID: 18039136
- PMCID: PMC2666336
- DOI: 10.1146/annurev.pathmechdis.3.121806.154305
Pancreatic cancer
Abstract
The past two decades have witnessed an explosion in our understanding of pancreatic cancer, and it is now clear that pancreatic cancer is a disease of inherited (germ-line) and somatic gene mutations. The genes mutated in pancreatic cancer include KRAS2, p16/CDKN2A, TP53, and SMAD4/DPC4, and these are accompanied by a substantial compendium of genomic and transcriptomic alterations that facilitate cell cycle deregulation, cell survival, invasion, and metastases. Pancreatic cancers do not arise de novo, and three distinct precursor lesions have been identified. Experimental models of pancreatic cancer have been developed in genetically engineered mice, which recapitulate the multistep progression of the cognate human disease. Although the putative cell of origin for pancreatic cancer remains elusive, minor populations of cells with stem-like properties have been identified that appear responsible for tumor initiation, metastases, and resistance of pancreatic cancer to conventional therapies.
Figures
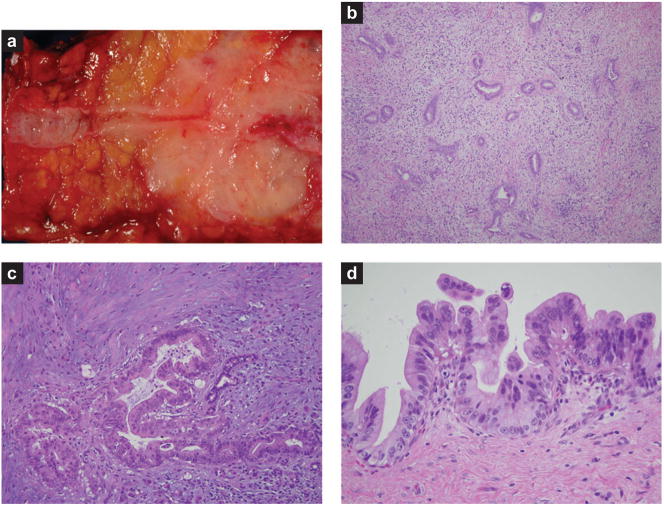
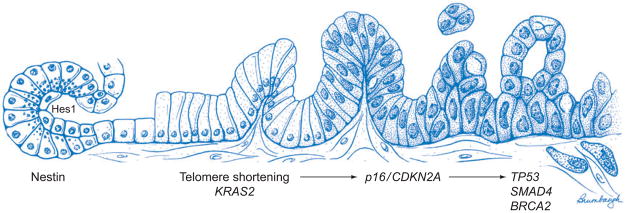
Similar articles
-
Immunohistochemically detected expression of 3 major genes (CDKN2A/p16, TP53, and SMAD4/DPC4) strongly predicts survival in patients with resectable pancreatic cancer.Ann Surg. 2013 Aug;258(2):336-46. doi: 10.1097/SLA.0b013e3182827a65. Ann Surg. 2013. PMID: 23470568
-
Molecular pathology of pancreatic cancer.Pathol Int. 2014 Jan;64(1):10-9. doi: 10.1111/pin.12114. Pathol Int. 2014. PMID: 24471965 Review.
-
Molecular genetics of pancreatic intraepithelial neoplasia.J Hepatobiliary Pancreat Surg. 2007;14(3):224-32. doi: 10.1007/s00534-006-1166-5. Epub 2007 May 29. J Hepatobiliary Pancreat Surg. 2007. PMID: 17520196 Free PMC article. Review.
-
KRAS2 mutations in human pancreatic acinar-ductal metaplastic lesions are limited to those with PanIN: implications for the human pancreatic cancer cell of origin.Mol Cancer Res. 2009 Feb;7(2):230-6. doi: 10.1158/1541-7786.MCR-08-0206. Epub 2009 Feb 10. Mol Cancer Res. 2009. PMID: 19208745 Free PMC article.
-
Genetic progression of pancreatic cancer.Cancer J. 2014 Jan-Feb;20(1):80-4. doi: 10.1097/PPO.0000000000000011. Cancer J. 2014. PMID: 24445769 Review.
Cited by
-
Circular RNA LIPH promotes pancreatic cancer glycolysis and progression through sponge miR-769-3p and interaction with GOLM1.Clin Transl Med. 2024 Aug;14(8):e70003. doi: 10.1002/ctm2.70003. Clin Transl Med. 2024. PMID: 39167076 Free PMC article. No abstract available.
-
Commitment and oncogene-induced plasticity of human stem cell-derived pancreatic acinar and ductal organoids.Cell Stem Cell. 2021 Jun 3;28(6):1090-1104.e6. doi: 10.1016/j.stem.2021.03.022. Epub 2021 Apr 28. Cell Stem Cell. 2021. PMID: 33915081 Free PMC article.
-
Proteogenomic insights into the biology and treatment of pancreatic ductal adenocarcinoma.J Hematol Oncol. 2022 Nov 25;15(1):168. doi: 10.1186/s13045-022-01384-3. J Hematol Oncol. 2022. PMID: 36434634 Free PMC article.
-
A Screen for Extracellular Signal-Regulated Kinase-Primed Glycogen Synthase Kinase 3 Substrates Identifies the p53 Inhibitor iASPP.J Virol. 2015 Sep;89(18):9232-41. doi: 10.1128/JVI.01072-15. Epub 2015 Jun 24. J Virol. 2015. PMID: 26109723 Free PMC article.
-
Targeted radionuclide therapies for pancreatic cancer.Cancer Gene Ther. 2015 Aug;22(8):375-9. doi: 10.1038/cgt.2015.32. Epub 2015 Jul 31. Cancer Gene Ther. 2015. PMID: 26227823 Free PMC article. Review.
References
-
- Am. Cancer Soc. Cancer Facts and Figures. Atlanta, GA: Am. Cancer Soc; 2007.
-
- Lowenfels AB, Maisonneuve P. Epidemiology and risk factors for pancreatic cancer. Best Pract Res Clin Gastroenterol. 2006;20:197–209. - PubMed
-
- Stolzenberg-Solomon RZ, Pietinen P, Barrett MJ, Taylor PR, Virtamo J, Albanes D. Dietary and other methyl-group availability factors and pancreatic cancer risk in a cohort of male smokers. Am J Epidemiol. 2001;153:680–87. - PubMed
-
- Villeneuve PJ, Johnson KC, Hanley AJ, Mao Y. Alcohol, tobacco and coffee consumption and the risk of pancreatic cancer: results from the Canadian Enhanced Surveillance System case-control project. Canadian Cancer Registries Epidemiology Research Group. Eur J Cancer Prev. 2000;9:49–58. - PubMed
-
- Michaud DS, Liu S, Giovannucci E, Willett WC, Colditz GA, Fuchs CS. Dietary sugar, glycemic load, and pancreatic cancer risk in a prospective study. J Natl Cancer Inst. 2002;94:1293–300. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
