

Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures
- PMID: 17994088
- PMCID: PMC2474711
- DOI: 10.1038/nature06340
Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures
Abstract
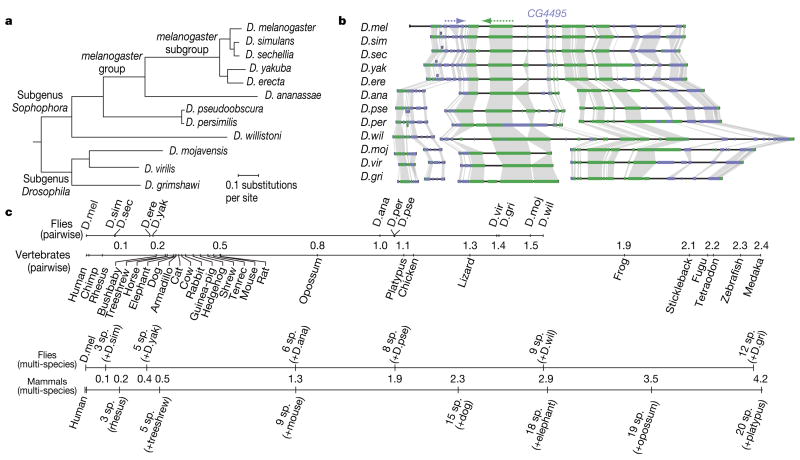
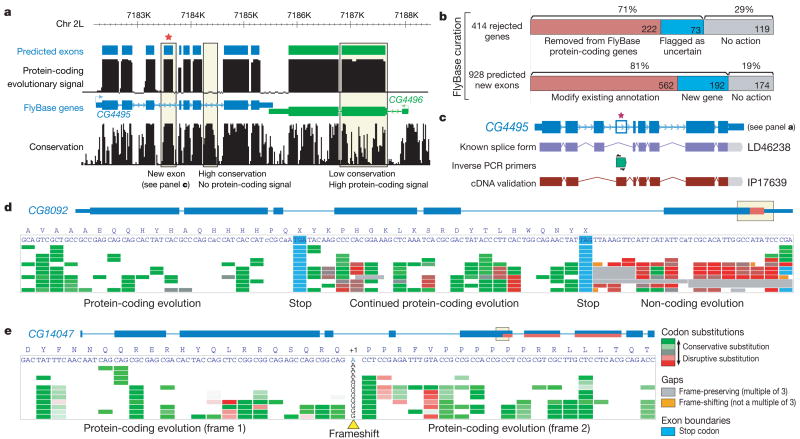
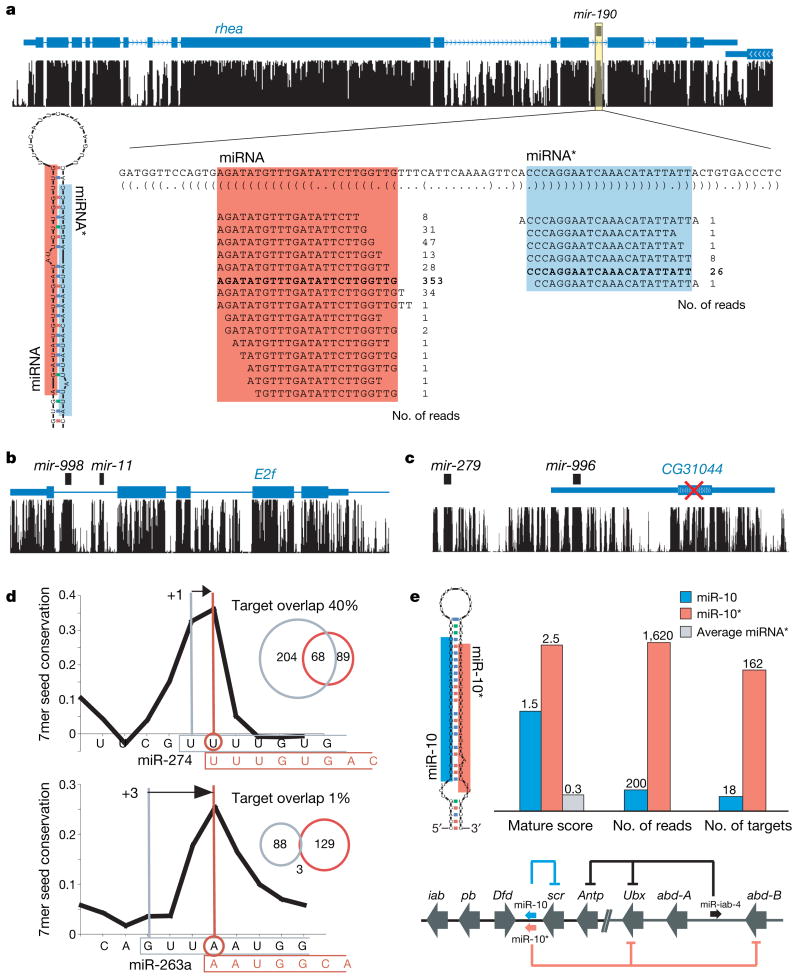
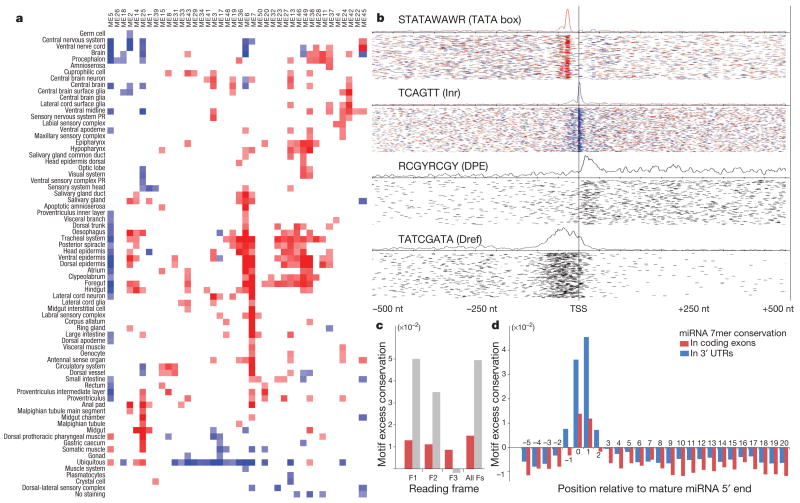
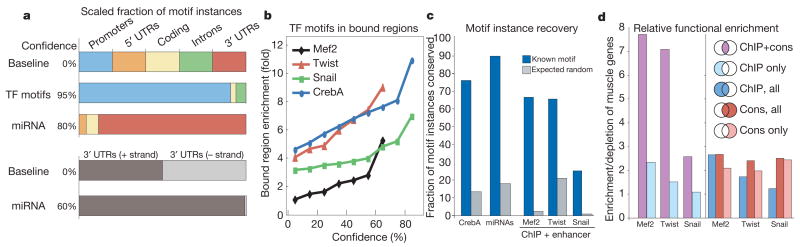
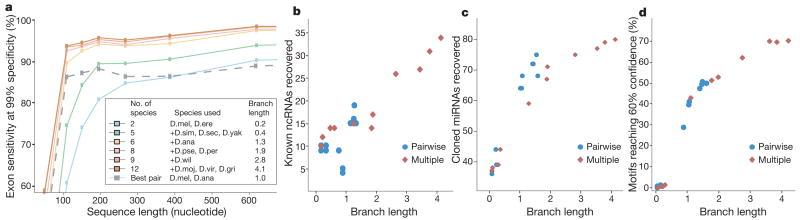
Sequencing of multiple related species followed by comparative genomics analysis constitutes a powerful approach for the systematic understanding of any genome. Here, we use the genomes of 12 Drosophila species for the de novo discovery of functional elements in the fly. Each type of functional element shows characteristic patterns of change, or 'evolutionary signatures', dictated by its precise selective constraints. Such signatures enable recognition of new protein-coding genes and exons, spurious and incorrect gene annotations, and numerous unusual gene structures, including abundant stop-codon readthrough. Similarly, we predict non-protein-coding RNA genes and structures, and new microRNA (miRNA) genes. We provide evidence of miRNA processing and functionality from both hairpin arms and both DNA strands. We identify several classes of pre- and post-transcriptional regulatory motifs, and predict individual motif instances with high confidence. We also study how discovery power scales with the divergence and number of species compared, and we provide general guidelines for comparative studies.
Figures


Comment in
-
Evolutionary genomics: come fly with us.Nature. 2007 Nov 8;450(7167):184-5. doi: 10.1038/450184a. Nature. 2007. PMID: 17994082 No abstract available.
Similar articles
-
Evolution of genes and genomes on the Drosophila phylogeny.Nature. 2007 Nov 8;450(7167):203-18. doi: 10.1038/nature06341. Nature. 2007. PMID: 17994087
-
Revisiting the protein-coding gene catalog of Drosophila melanogaster using 12 fly genomes.Genome Res. 2007 Dec;17(12):1823-36. doi: 10.1101/gr.6679507. Epub 2007 Nov 7. Genome Res. 2007. PMID: 17989253 Free PMC article.
-
Systematic discovery and characterization of fly microRNAs using 12 Drosophila genomes.Genome Res. 2007 Dec;17(12):1865-79. doi: 10.1101/gr.6593807. Epub 2007 Nov 7. Genome Res. 2007. PMID: 17989255 Free PMC article.
-
Regulatory RNAs in the light of Drosophila genomics.Brief Funct Genomics. 2012 Sep;11(5):356-65. doi: 10.1093/bfgp/els033. Epub 2012 Sep 5. Brief Funct Genomics. 2012. PMID: 22956639 Free PMC article. Review.
-
Helitrons shaping the genomic architecture of Drosophila: enrichment of DINE-TR1 in α- and β-heterochromatin, satellite DNA emergence, and piRNA expression.Chromosome Res. 2015 Sep;23(3):597-613. doi: 10.1007/s10577-015-9480-x. Chromosome Res. 2015. PMID: 26408292 Review.
Cited by
-
Prediction of clustered RNA-binding protein motif sites in the mammalian genome.Nucleic Acids Res. 2013 Aug;41(14):6793-807. doi: 10.1093/nar/gkt421. Epub 2013 May 18. Nucleic Acids Res. 2013. PMID: 23685613 Free PMC article.
-
Preferential regulation of stably expressed genes in the human genome suggests a widespread expression buffering role of microRNAs.BMC Genomics. 2012;13 Suppl 7(Suppl 7):S14. doi: 10.1186/1471-2164-13-S7-S14. Epub 2012 Dec 13. BMC Genomics. 2012. PMID: 23281599 Free PMC article.
-
miRmap web: Comprehensive microRNA target prediction online.Nucleic Acids Res. 2013 Jul;41(Web Server issue):W165-8. doi: 10.1093/nar/gkt430. Epub 2013 May 28. Nucleic Acids Res. 2013. PMID: 23716633 Free PMC article.
-
Targeting microRNAs in cancer: rationale, strategies and challenges.Nat Rev Drug Discov. 2010 Oct;9(10):775-89. doi: 10.1038/nrd3179. Nat Rev Drug Discov. 2010. PMID: 20885409 Free PMC article. Review.
-
Fine-tuning enhancer models to predict transcriptional targets across multiple genomes.PLoS One. 2007 Nov 7;2(11):e1115. doi: 10.1371/journal.pone.0001115. PLoS One. 2007. PMID: 17973026 Free PMC article.
References
-
- Miller W, Makova KD, Nekrutenko A, Hardison RC. Comparative genomics. Annu Rev Genomics Hum Genet. 2004;5:15–56. - PubMed
-
- Ureta-Vidal A, Ettwiller L, Birney E. Comparative genomics: genome-wide analysis in metazoan eukaryotes. Nature Rev Genet. 2003;4:251–262. - PubMed
-
- Kellis M, et al. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423:241–254. - PubMed
-
- Cliften P, et al. Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science. 2003;301:71–76. - PubMed
-
- Brent MR. Genome annotation past, present, and future: how to define an ORF at each locus. Genome Res. 2005;15:1777–1786. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
