

Gefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics
- PMID: 17973573
- PMCID: PMC2043013
- DOI: 10.1371/journal.pmed.0040316
Gefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics
Abstract
Background: The epidermal growth factor receptor (EGFR) plays a critical role in the control of cellular proliferation, differentiation, and survival. Abnormalities in EGF-EGFR signaling, such as mutations that render the EGFR hyperactive or cause overexpression of the wild-type receptor, have been found in a broad range of cancers, including carcinomas of the lung, breast, and colon. EGFR inhibitors such as gefitinib have proven successful in the treatment of certain cancers, particularly non-small cell lung cancers (NSCLCs) harboring activating mutations within the EGFR gene, but the molecular mechanisms leading to tumor regression remain unknown. Therefore, we wished to delineate these mechanisms.
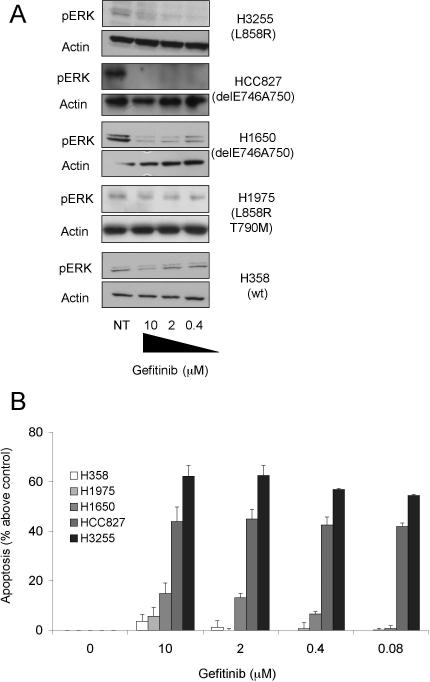
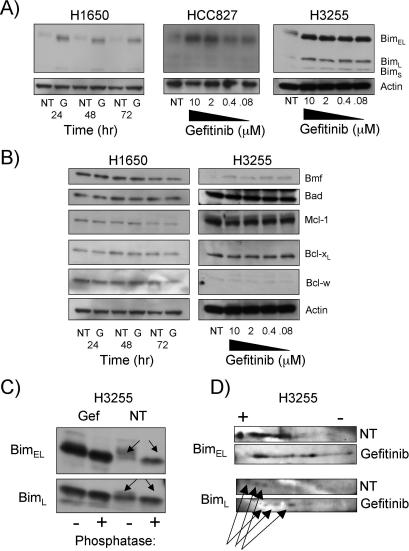
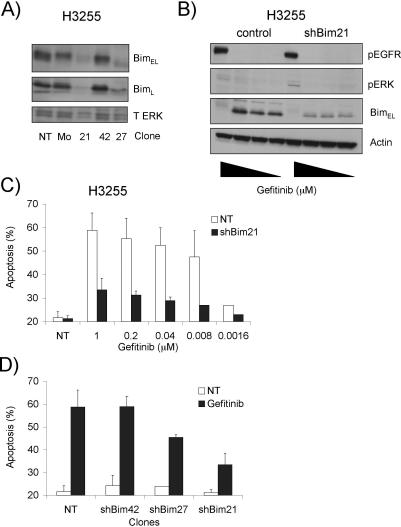
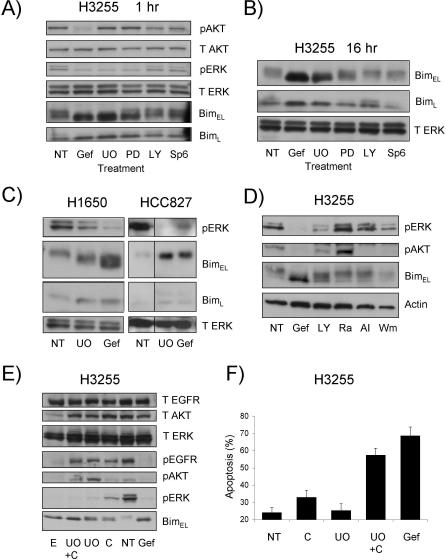
Methods and findings: We performed biochemical and genetic studies to investigate the mechanisms by which inhibitors of EGFR tyrosine kinase activity, such as gefitinib, inhibit the growth of human NSCLCs. We found that gefitinib triggered intrinsic (also called "mitochondrial") apoptosis signaling, involving the activation of BAX and mitochondrial release of cytochrome c, ultimately unleashing the caspase cascade. Gefitinib caused a rapid increase in the level of the proapoptotic BH3-only protein BIM (also called BCL2-like 11) through both transcriptional and post-translational mechanisms. Experiments with pharmacological inhibitors indicated that blockade of MEK-ERK1/2 (mitogen-activated protein kinase kinase-extracellular signal-regulated protein kinase 1/2) signaling, but not blockade of PI3K (phosphatidylinositol 3-kinase), JNK (c-Jun N-terminal kinase or mitogen-activated protein kinase 8), or AKT (protein kinase B), was critical for BIM activation. Using RNA interference, we demonstrated that BIM is essential for gefitinib-induced killing of NSCLC cells. Moreover, we found that gefitinib-induced apoptosis is enhanced by addition of the BH3 mimetic ABT-737.
Conclusions: Inhibitors of the EGFR tyrosine kinase have proven useful in the therapy of certain cancers, in particular NSCLCs possessing activating mutations in the EGFR kinase domain, but the mechanisms of tumor cell killing are still unclear. In this paper, we demonstrate that activation of the proapoptotic BH3-only protein BIM is essential for tumor cell killing and that shutdown of the EGFR-MEK-ERK signaling cascade is critical for BIM activation. Moreover, we demonstrate that addition of a BH3 mimetic significantly enhances killing of NSCLC cells by the EGFR tyrosine kinase inhibitor gefitinib. It appears likely that this approach represents a paradigm shared by many, and perhaps all, oncogenic tyrosine kinases and suggests a powerful new strategy for cancer therapy.
Conflict of interest statement
Figures

Similar articles
-
BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations.PLoS Med. 2007 Oct;4(10):1669-79; discussion 1680. doi: 10.1371/journal.pmed.0040315. PLoS Med. 2007. PMID: 17973572 Free PMC article.
-
PUMA and BIM are required for oncogene inactivation-induced apoptosis.Sci Signal. 2013 Mar 26;6(268):ra20. doi: 10.1126/scisignal.2003483. Sci Signal. 2013. PMID: 23532334 Free PMC article.
-
Role of survivin in EGFR inhibitor-induced apoptosis in non-small cell lung cancers positive for EGFR mutations.Cancer Res. 2010 Dec 15;70(24):10402-10. doi: 10.1158/0008-5472.CAN-10-2438. Cancer Res. 2010. PMID: 21159653
-
Clinical significance of epidermal growth factor receptor tyrosine kinase inhibitors: sensitivity and resistance.Respir Investig. 2014 Nov;52(6):348-56. doi: 10.1016/j.resinv.2014.10.002. Epub 2014 Oct 31. Respir Investig. 2014. PMID: 25453378 Review.
-
EGF receptor in relation to tumor development: molecular basis of responsiveness of cancer cells to EGFR-targeting tyrosine kinase inhibitors.FEBS J. 2010 Jan;277(2):316-26. doi: 10.1111/j.1742-4658.2009.07450.x. Epub 2009 Nov 18. FEBS J. 2010. PMID: 19922467 Review.
Cited by
-
Primary Double-Strike Therapy for Cancers to Overcome EGFR Kinase Inhibitor Resistance: Proposal from the Bench.J Thorac Oncol. 2017 Jan;12(1):27-35. doi: 10.1016/j.jtho.2016.09.003. Epub 2016 Sep 15. J Thorac Oncol. 2017. PMID: 27642065 Free PMC article. Review.
-
BIM expression in treatment-naive cancers predicts responsiveness to kinase inhibitors.Cancer Discov. 2011 Sep;1(4):352-65. doi: 10.1158/2159-8290.CD-11-0106. Epub 2011 Jul 22. Cancer Discov. 2011. PMID: 22145099 Free PMC article.
-
Hepatocyte growth factor renders BRAF mutant human melanoma cell lines resistant to PLX4032 by downregulating the pro-apoptotic BH3-only proteins PUMA and BIM.Cell Death Differ. 2016 Dec;23(12):2054-2062. doi: 10.1038/cdd.2016.96. Epub 2016 Sep 30. Cell Death Differ. 2016. PMID: 27689874 Free PMC article.
-
Adapted to Survive: Targeting Cancer Cells with BH3 Mimetics.Cancer Discov. 2022 May 2;12(5):1217-1232. doi: 10.1158/2159-8290.CD-21-1334. Cancer Discov. 2022. PMID: 35491624 Free PMC article. Review.
-
Pharmaceutical Reactivation of Attenuated Apoptotic Pathways Leads to Elimination of Osimertinib Drug-Tolerant Cells.Cancer Res Commun. 2022 Oct 31;2(10):1312-1325. doi: 10.1158/2767-9764.CRC-22-0066. eCollection 2022 Oct. Cancer Res Commun. 2022. PMID: 36969743 Free PMC article.
References
-
- Mendelsohn J, Baselga J. Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J Clin Oncol. 2003;21:2787–2799. - PubMed
-
- Ji H, Li D, Chen L, Shimamura T, Kobayashi S, et al. The impact of human EGFR kinase domain mutations on lung tumorigenesis and in vivo sensitivity to EGFR-targeted therapies. Cancer Cell. 2006;9:485–495. - PubMed
-
- Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
