

Models, mechanisms and clinical evidence for cancer dormancy
- PMID: 17957189
- PMCID: PMC2519109
- DOI: 10.1038/nrc2256
Models, mechanisms and clinical evidence for cancer dormancy
Abstract
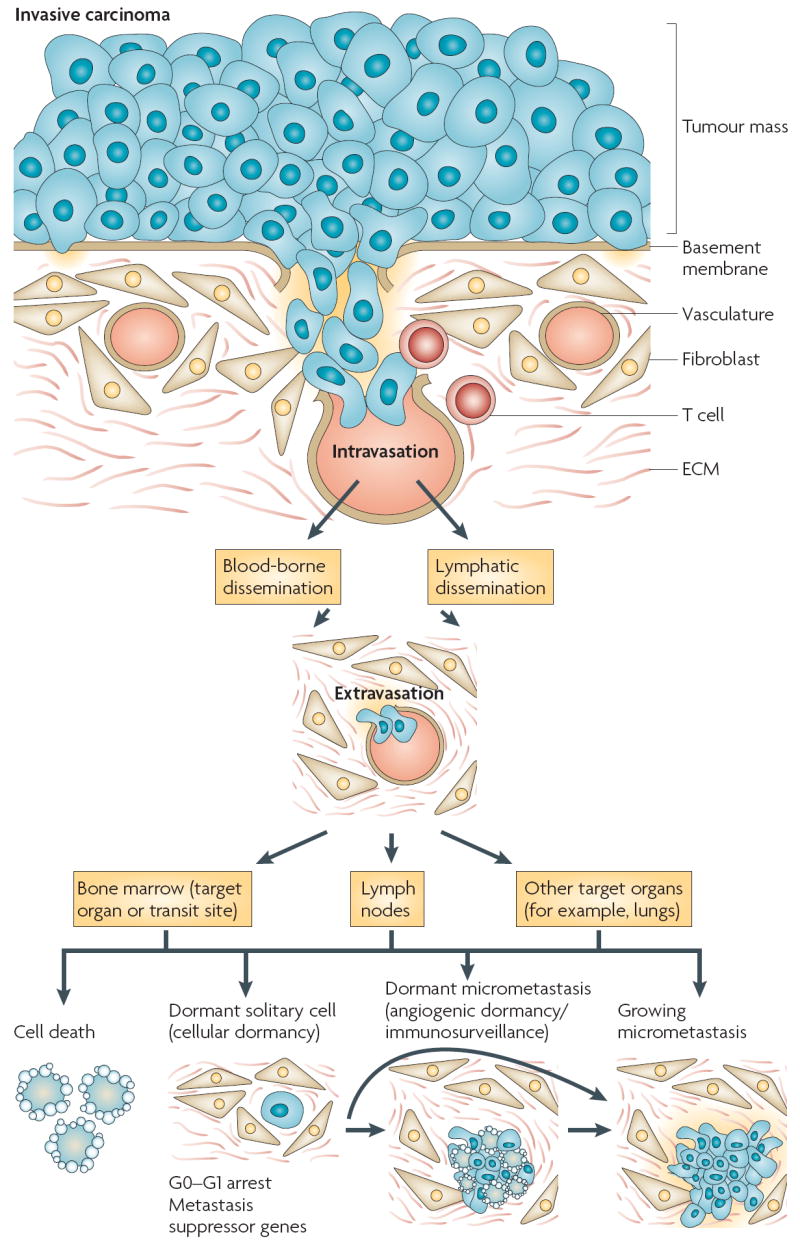
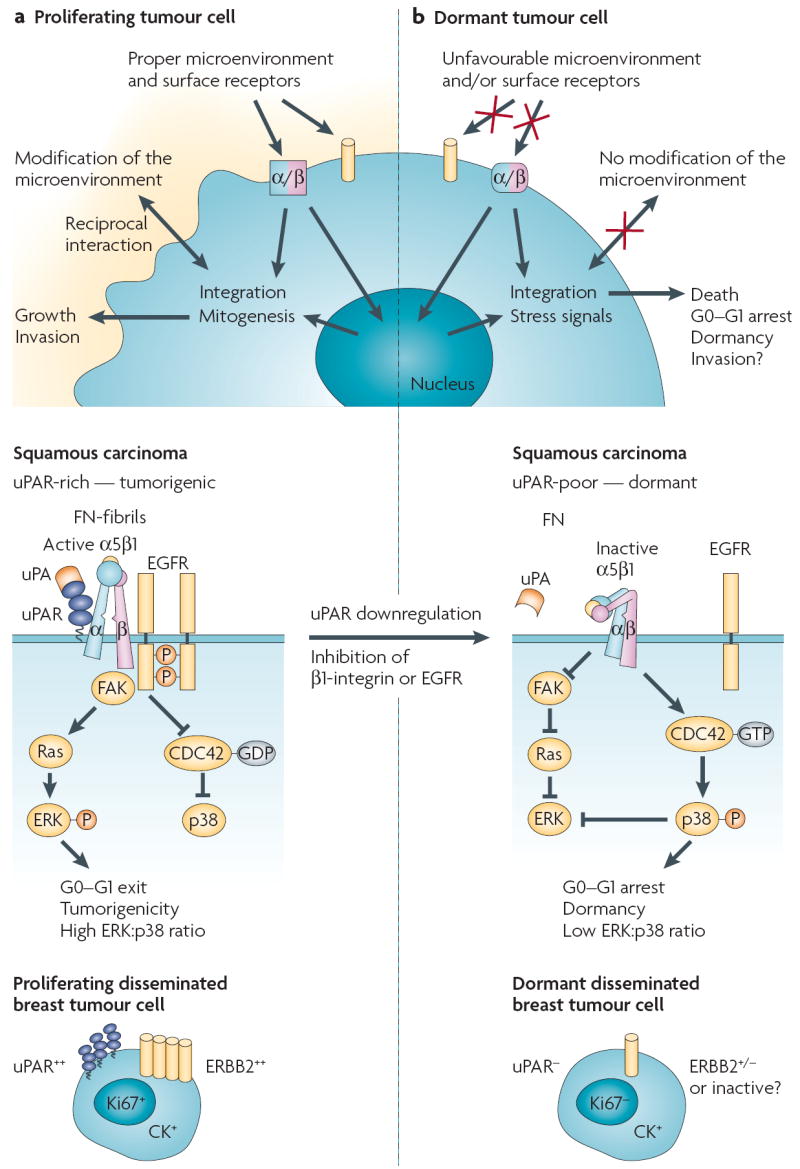
Patients with cancer can develop recurrent metastatic disease with latency periods that range from years even to decades. This pause can be explained by cancer dormancy, a stage in cancer progression in which residual disease is present but remains asymptomatic. Cancer dormancy is poorly understood, resulting in major shortcomings in our understanding of the full complexity of the disease. Here, I review experimental and clinical evidence that supports the existence of various mechanisms of cancer dormancy including angiogenic dormancy, cellular dormancy (G0-G1 arrest) and immunosurveillance. The advances in this field provide an emerging picture of how cancer dormancy can ensue and how it could be therapeutically targeted.
Figures


Similar articles
-
Cellular dormancy in minimal residual disease following targeted therapy.Breast Cancer Res. 2021 Jun 4;23(1):63. doi: 10.1186/s13058-021-01416-9. Breast Cancer Res. 2021. PMID: 34088357 Free PMC article.
-
Tumor dormancy and cancer stem cells: two sides of the same coin?Adv Exp Med Biol. 2013;734:145-79. doi: 10.1007/978-1-4614-1445-2_8. Adv Exp Med Biol. 2013. PMID: 23143979
-
[Tumor dormancy and identification of therapeutic targets].Ai Zheng. 2009 May;28(5):555-8. Ai Zheng. 2009. PMID: 19624889 Review. Chinese.
-
Tumor dormancy of primary and secondary cancers.APMIS. 2008 Jul-Aug;116(7-8):615-28. doi: 10.1111/j.1600-0463.2008.01077.x. APMIS. 2008. PMID: 18834406 Review.
-
The sweet and sour of cancer: glycans as novel therapeutic targets.Nat Rev Cancer. 2005 Jul;5(7):526-42. doi: 10.1038/nrc1649. Nat Rev Cancer. 2005. PMID: 16069816 Review.
Cited by
-
Caught in the act: revealing the metastatic process by live imaging.Dis Model Mech. 2013 May;6(3):580-93. doi: 10.1242/dmm.009282. Dis Model Mech. 2013. PMID: 23616077 Free PMC article. Review.
-
Evolutionary determinants of cancer.Cancer Discov. 2015 Aug;5(8):806-20. doi: 10.1158/2159-8290.CD-15-0439. Epub 2015 Jul 20. Cancer Discov. 2015. PMID: 26193902 Free PMC article. Review.
-
The diverse and complex roles of radiation on cancer treatment: therapeutic target and genome maintenance.Am J Cancer Res. 2012;2(4):372-82. Epub 2012 Jun 28. Am J Cancer Res. 2012. PMID: 22860229 Free PMC article.
-
Slow-cycling (dormant) cancer cells in therapy resistance, cancer relapse and metastasis.Semin Cancer Biol. 2022 Jan;78:90-103. doi: 10.1016/j.semcancer.2021.04.021. Epub 2021 May 9. Semin Cancer Biol. 2022. PMID: 33979674 Free PMC article. Review.
-
Clonal evolution in cancer.Nature. 2012 Jan 18;481(7381):306-13. doi: 10.1038/nature10762. Nature. 2012. PMID: 22258609 Free PMC article. Review.
References
-
- Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nature Rev Cancer. 2004;4:448–456. - PubMed
-
-
Schmidt-Kittler O, et al. From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci U S A. 2003;100:7737–7742.First evidence that breast cancer cells can disseminate in a far less progressed genomic state than previously thought. Genomic aberrations that make them metastatic are acquired after this step.
-
-
- Karrison TG, Ferguson DJ, Meier P. Dormancy of mammary carcinoma after mastectomy. J Natl Cancer Inst. 1999;91:80–85. - PubMed
-
- Pfitzenmaier J, et al. Telomerase activity in disseminated prostate cancer cells. BJU Int. 2006;97:1309–1313. - PubMed
-
- Weckermann D, et al. Disseminated cytokeratin positive tumor cells in the bone marrow of patients with prostate cancer: detection and prognostic value. J Urol. 2001;166:699–703. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
