

Current approaches to gene regulatory network modelling
- PMID: 17903290
- PMCID: PMC1995542
- DOI: 10.1186/1471-2105-8-S6-S9
Current approaches to gene regulatory network modelling
Abstract
Many different approaches have been developed to model and simulate gene regulatory networks. We proposed the following categories for gene regulatory network models: network parts lists, network topology models, network control logic models, and dynamic models. Here we will describe some examples for each of these categories. We will study the topology of gene regulatory networks in yeast in more detail, comparing a direct network derived from transcription factor binding data and an indirect network derived from genome-wide expression data in mutants. Regarding the network dynamics we briefly describe discrete and continuous approaches to network modelling, then describe a hybrid model called Finite State Linear Model and demonstrate that some simple network dynamics can be simulated in this model.
Figures
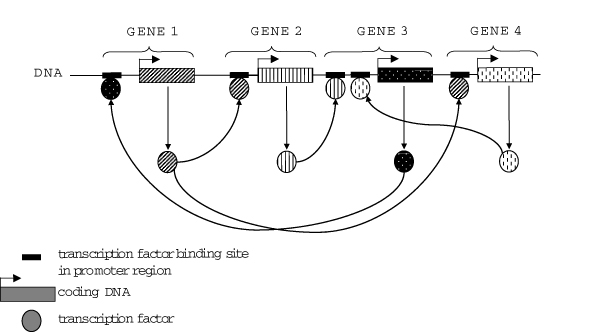
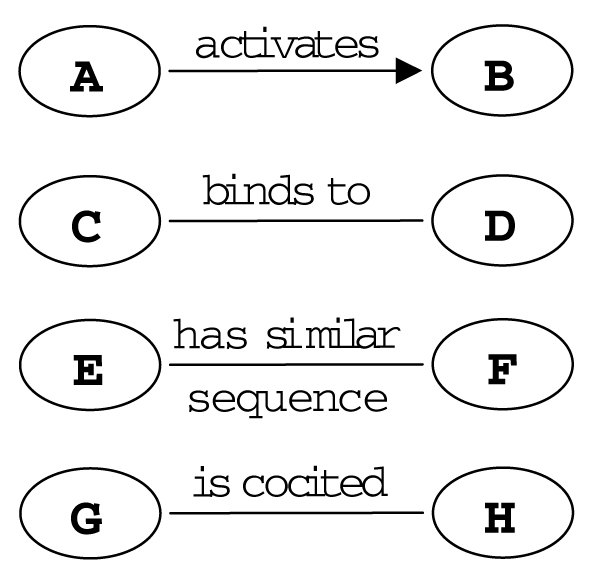

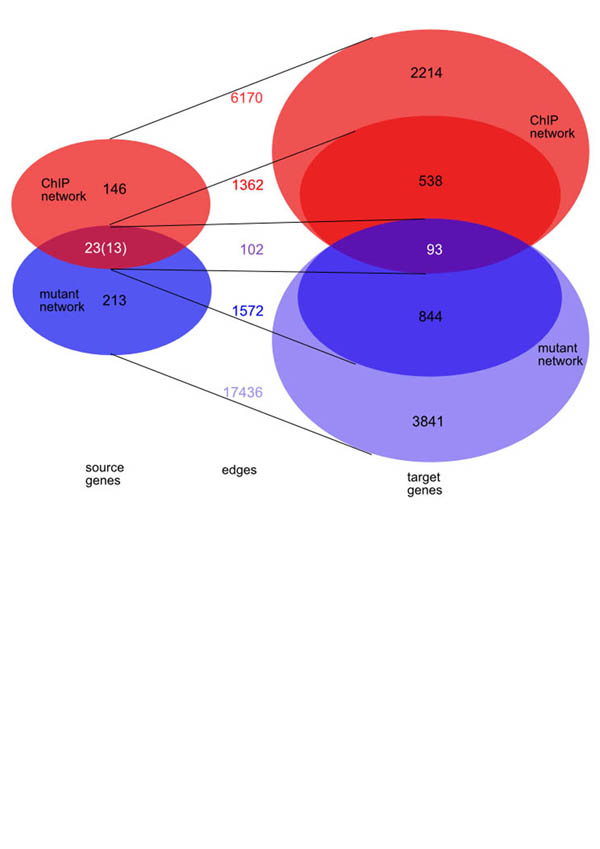

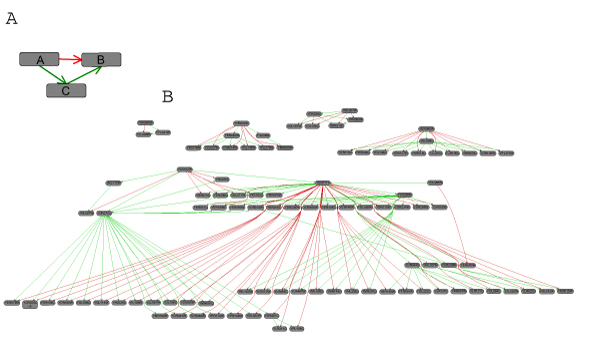
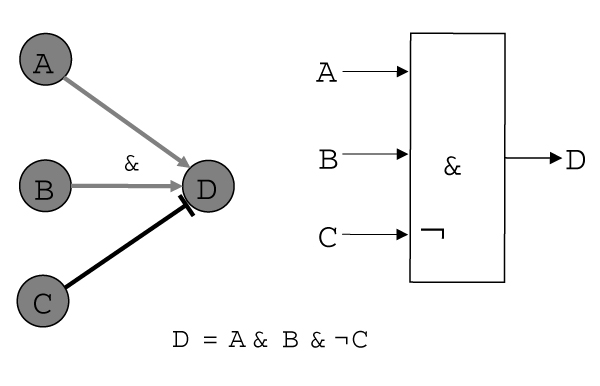

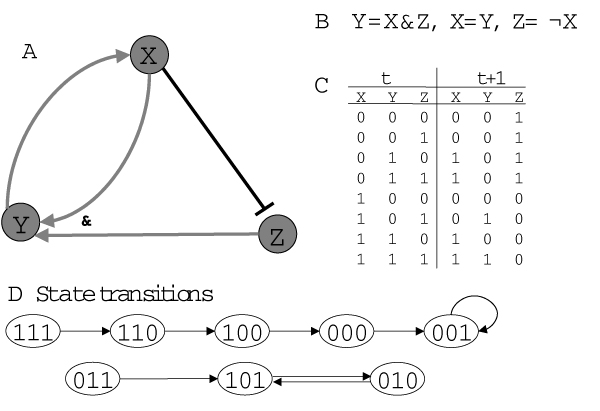


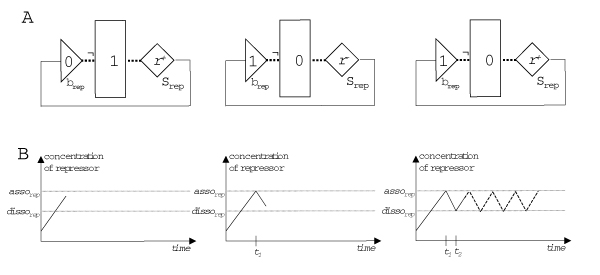


Republished from
-
Modelling gene networks at different organisational levels.FEBS Lett. 2005 Mar 21;579(8):1859-66. doi: 10.1016/j.febslet.2005.01.073. FEBS Lett. 2005. PMID: 15763564 Review.
-
Modelling in molecular biology: describing transcription regulatory networks at different scales.Philos Trans R Soc Lond B Biol Sci. 2006 Mar 29;361(1467):483-94. doi: 10.1098/rstb.2005.1806. Philos Trans R Soc Lond B Biol Sci. 2006. PMID: 16524837 Free PMC article. Review.
Similar articles
-
Modelling in molecular biology: describing transcription regulatory networks at different scales.Philos Trans R Soc Lond B Biol Sci. 2006 Mar 29;361(1467):483-94. doi: 10.1098/rstb.2005.1806. Philos Trans R Soc Lond B Biol Sci. 2006. PMID: 16524837 Free PMC article. Review.
-
Modelling gene networks at different organisational levels.FEBS Lett. 2005 Mar 21;579(8):1859-66. doi: 10.1016/j.febslet.2005.01.073. FEBS Lett. 2005. PMID: 15763564 Review.
-
Comparing different ODE modelling approaches for gene regulatory networks.J Theor Biol. 2009 Dec 21;261(4):511-30. doi: 10.1016/j.jtbi.2009.07.040. Epub 2009 Aug 6. J Theor Biol. 2009. PMID: 19665034
-
Mosaic gene network modelling identified new regulatory mechanisms in HCV infection.Virus Res. 2016 Jun 15;218:71-8. doi: 10.1016/j.virusres.2015.10.004. Epub 2015 Oct 22. Virus Res. 2016. PMID: 26481968
-
Dynamics of gene regulatory networks and their dependence on network topology and quantitative parameters - the case of phage λ.BMC Bioinformatics. 2019 May 31;20(1):296. doi: 10.1186/s12859-019-2909-z. BMC Bioinformatics. 2019. PMID: 31151381 Free PMC article.
Cited by
-
SuMO-Fil: Supervised multi-omic filtering prior to performing network analysis.PLoS One. 2021 Aug 3;16(8):e0255579. doi: 10.1371/journal.pone.0255579. eCollection 2021. PLoS One. 2021. PMID: 34343218 Free PMC article.
-
Neurogenetic and Neuroepigenetic Mechanisms in Cognitive Health and Disease.Front Mol Neurosci. 2020 Dec 3;13:205. doi: 10.3389/fnmol.2020.589109. eCollection 2020. Front Mol Neurosci. 2020. PMID: 33343294 Free PMC article.
-
Elucidation of functional consequences of signalling pathway interactions.BMC Bioinformatics. 2009 Nov 6;10:370. doi: 10.1186/1471-2105-10-370. BMC Bioinformatics. 2009. PMID: 19895694 Free PMC article.
-
Boolean modelling as a logic-based dynamic approach in systems medicine.Comput Struct Biotechnol J. 2022 Jun 17;20:3161-3172. doi: 10.1016/j.csbj.2022.06.035. eCollection 2022. Comput Struct Biotechnol J. 2022. PMID: 35782730 Free PMC article. Review.
-
Modeling formalisms in Systems Biology.AMB Express. 2011 Dec 5;1:45. doi: 10.1186/2191-0855-1-45. AMB Express. 2011. PMID: 22141422 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
