

Genomics of Actinobacteria: tracing the evolutionary history of an ancient phylum
- PMID: 17804669
- PMCID: PMC2168647
- DOI: 10.1128/MMBR.00005-07
Genomics of Actinobacteria: tracing the evolutionary history of an ancient phylum
Abstract
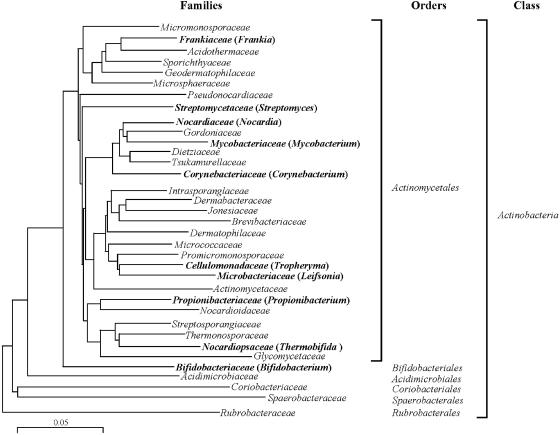
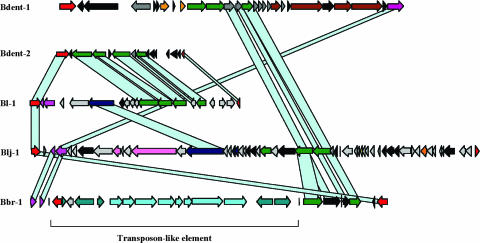
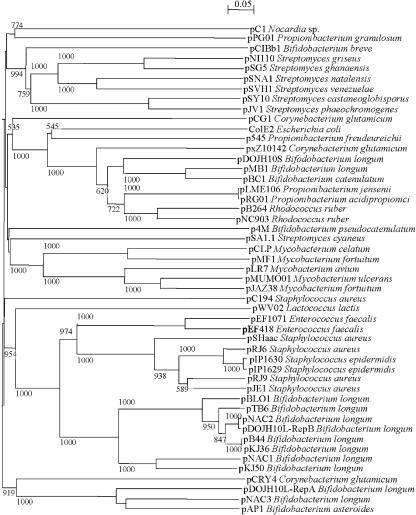
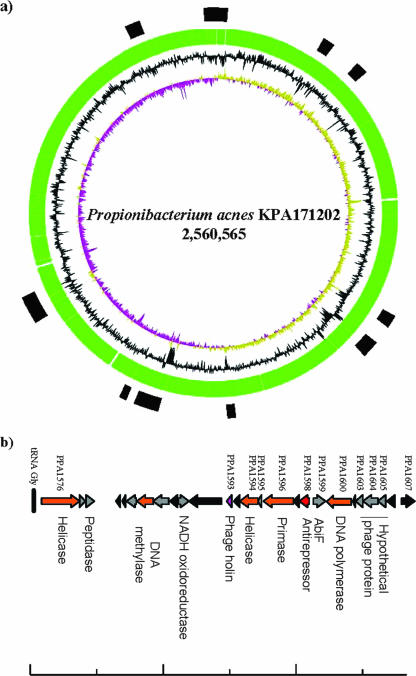
Actinobacteria constitute one of the largest phyla among bacteria and represent gram-positive bacteria with a high G+C content in their DNA. This bacterial group includes microorganisms exhibiting a wide spectrum of morphologies, from coccoid to fragmenting hyphal forms, as well as possessing highly variable physiological and metabolic properties. Furthermore, Actinobacteria members have adopted different lifestyles, and can be pathogens (e.g., Corynebacterium, Mycobacterium, Nocardia, Tropheryma, and Propionibacterium), soil inhabitants (Streptomyces), plant commensals (Leifsonia), or gastrointestinal commensals (Bifidobacterium). The divergence of Actinobacteria from other bacteria is ancient, making it impossible to identify the phylogenetically closest bacterial group to Actinobacteria. Genome sequence analysis has revolutionized every aspect of bacterial biology by enhancing the understanding of the genetics, physiology, and evolutionary development of bacteria. Various actinobacterial genomes have been sequenced, revealing a wide genomic heterogeneity probably as a reflection of their biodiversity. This review provides an account of the recent explosion of actinobacterial genomics data and an attempt to place this in a biological and evolutionary context.
Figures






Similar articles
-
Taxonomy, Physiology, and Natural Products of Actinobacteria.Microbiol Mol Biol Rev. 2015 Nov 25;80(1):1-43. doi: 10.1128/MMBR.00019-15. Print 2016 Mar. Microbiol Mol Biol Rev. 2015. PMID: 26609051 Free PMC article. Review.
-
Distribution and Evolutionary History of Sialic Acid Catabolism in the Phylum Actinobacteria.Microbiol Spectr. 2022 Feb 23;10(1):e0238021. doi: 10.1128/spectrum.02380-21. Epub 2022 Jan 12. Microbiol Spectr. 2022. PMID: 35019771 Free PMC article.
-
Signature proteins that are distinctive characteristics of Actinobacteria and their subgroups.Antonie Van Leeuwenhoek. 2006 Jul;90(1):69-91. doi: 10.1007/s10482-006-9061-2. Epub 2006 May 3. Antonie Van Leeuwenhoek. 2006. PMID: 16670965
-
Comparative analysis of Mycobacterium and related Actinomycetes yields insight into the evolution of Mycobacterium tuberculosis pathogenesis.BMC Genomics. 2012 Mar 28;13:120. doi: 10.1186/1471-2164-13-120. BMC Genomics. 2012. PMID: 22452820 Free PMC article.
-
Genomics as a means to understand bacterial phylogeny and ecological adaptation: the case of bifidobacteria.Antonie Van Leeuwenhoek. 2007 May;91(4):351-72. doi: 10.1007/s10482-006-9122-6. Epub 2006 Oct 28. Antonie Van Leeuwenhoek. 2007. PMID: 17072531 Review.
Cited by
-
A two-component regulatory system controls autoregulated serpin expression in Bifidobacterium breve UCC2003.Appl Environ Microbiol. 2012 Oct;78(19):7032-41. doi: 10.1128/AEM.01776-12. Epub 2012 Jul 27. Appl Environ Microbiol. 2012. PMID: 22843530 Free PMC article.
-
Proteome turnover in bacteria: current status for Corynebacterium glutamicum and related bacteria.Microb Biotechnol. 2013 Nov;6(6):708-19. doi: 10.1111/1751-7915.12035. Epub 2013 Feb 20. Microb Biotechnol. 2013. PMID: 23425033 Free PMC article. Review.
-
Phylogenetic framework and molecular signatures for the main clades of the phylum Actinobacteria.Microbiol Mol Biol Rev. 2012 Mar;76(1):66-112. doi: 10.1128/MMBR.05011-11. Microbiol Mol Biol Rev. 2012. PMID: 22390973 Free PMC article. Review.
-
Corynebacterium urealyticum: a comprehensive review of an understated organism.Infect Drug Resist. 2015 May 21;8:129-45. doi: 10.2147/IDR.S74795. eCollection 2015. Infect Drug Resist. 2015. PMID: 26056481 Free PMC article. Review.
-
Bifidobacterium asteroides PRL2011 genome analysis reveals clues for colonization of the insect gut.PLoS One. 2012;7(9):e44229. doi: 10.1371/journal.pone.0044229. Epub 2012 Sep 20. PLoS One. 2012. PMID: 23028506 Free PMC article.
References
-
- Aderem, A. 2005. Systems biology: its practice and challenges. Cell 121:511-513. - PubMed
-
- Adindla, S., and L. Guruprasad. 2003. Sequence analysis corresponding to the PPE and PE proteins in Mycobacterium tuberculosis and other genomes. J. Biosci. 28:169-179. - PubMed
-
- Alam, M. S., S. K. Garg, and P. Agrawal. 2007. Molecular function of WhiB4/Rv3681c of Mycobacterium tuberculosis H37Rv: a [4Fe-4S] cluster co-ordinating protein disulphide reductase. Mol. Microbiol. 63:1414-1431. - PubMed
-
- Alvarez-Martin, P., A. B. Florez, and B. Mayo. 2006. Screening for plasmids among human bifidobacteria species: sequencing and analysis of pBC1 from Bifidobacterium catenulatum L48. Plasmid 57:165-174. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
