

Differential regulation of class IA phosphoinositide 3-kinase catalytic subunits p110 alpha and beta by protease-activated receptor 2 and beta-arrestins
- PMID: 17680774
- PMCID: PMC2267348
- DOI: 10.1042/BJ20070483
Differential regulation of class IA phosphoinositide 3-kinase catalytic subunits p110 alpha and beta by protease-activated receptor 2 and beta-arrestins
Abstract
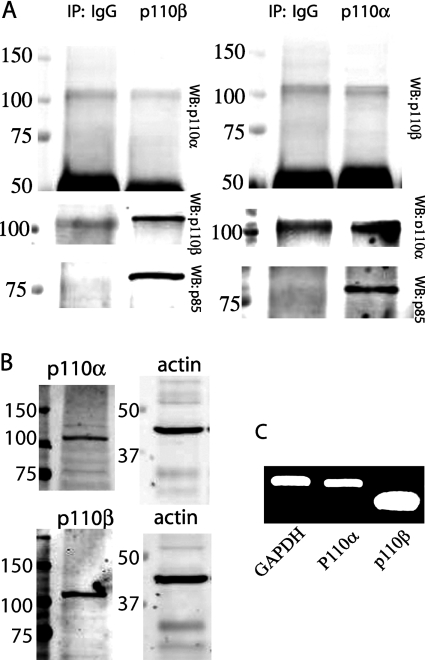
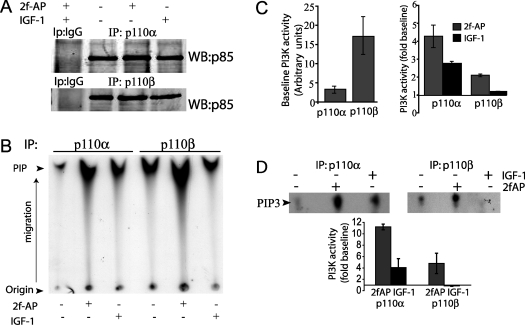
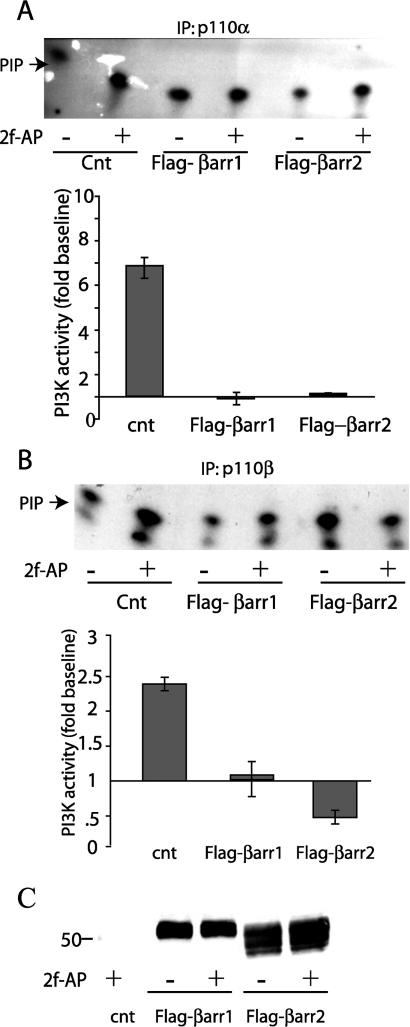
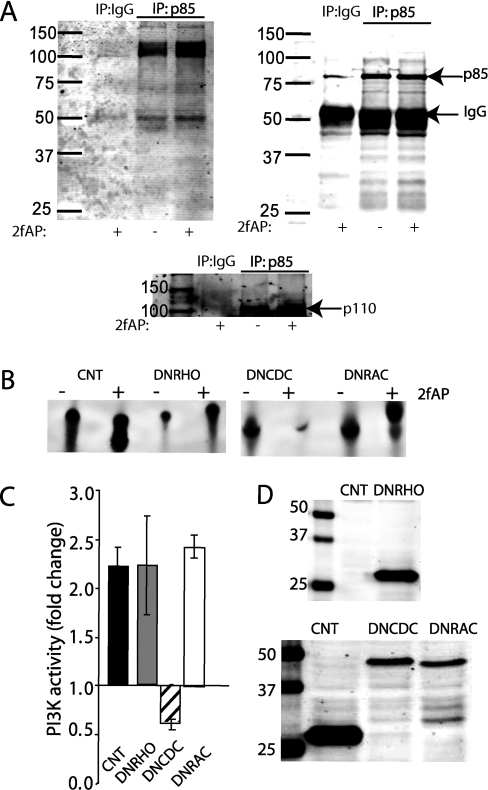
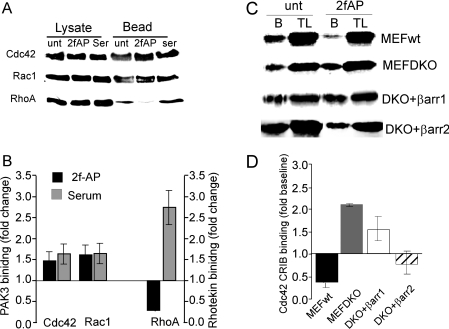
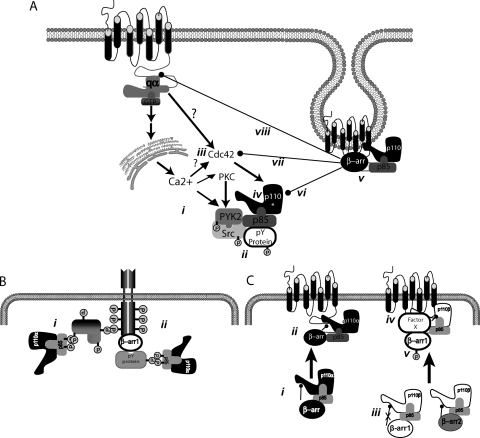
PAR-2 (protease-activated receptor 2) is a GPCR (G-protein-coupled receptor) that can elicit both G-protein-dependent and -independent signals. We have shown previously that PAR-2 simultaneously promotes Galphaq/Ca2+-dependent activation and beta-arrestin-1-dependent inhibition of class IA PI3K (phosphoinositide 3-kinase), and we sought to characterize further the role of beta-arrestins in the regulation of PI3K activity. Whereas the ability of beta-arrestin-1 to inhibit p110alpha (PI3K catalytic subunit alpha) has been demonstrated, the role of beta-arrestin-2 in PI3K regulation and possible differences in the regulation of the two catalytic subunits (p110alpha and p110beta) associated with p85alpha (PI3K regulatory subunit) have not been examined. In the present study we have demonstrated that: (i) PAR-2 increases p110alpha- and p110beta-associated lipid kinase activities, and both p110alpha and p110beta are inhibited by over-expression of either beta-arrestin-1 or -2; (ii) both beta-arrestin-1 and -2 directly inhibit the p110alpha catalytic subunit in vitro, whereas only beta-arrestin-2 directly inhibited p110beta; (iii) examination of upstream pathways revealed that PAR-2-induced PI3K activity required the small GTPase Cdc (cell-division cycle)42, but not tyrosine phosphorylation of p85; and (iv) beta-arrestins inhibit PAR-2-induced Cdc42 activation. Taken together, these results indicated that beta-arrestins could inhibit PAR-2-stimulated PI3K activity, both directly and through interference with upstream pathways, and that the two beta-arrestins differ in their ability to inhibit the p110alpha and p110beta catalytic subunits. These results are particularly important in light of the growing interest in PAR-2 as a pharmacological target, as commonly used biochemical assays that monitor G-protein coupling would not screen for beta-arrestin-dependent signalling events.
Figures

Similar articles
-
Protease-activated receptor-2 simultaneously directs beta-arrestin-1-dependent inhibition and Galphaq-dependent activation of phosphatidylinositol 3-kinase.Biochemistry. 2006 Aug 8;45(31):9374-85. doi: 10.1021/bi0602617. Biochemistry. 2006. PMID: 16878972
-
PAR-1 activation rescues astrocytes through the PI3K/Akt signaling pathway from chemically induced apoptosis that is exacerbated by gene silencing of β-arrestin 1.Neurochem Int. 2014 Feb;67:46-56. doi: 10.1016/j.neuint.2013.12.007. Epub 2013 Dec 27. Neurochem Int. 2014. PMID: 24378649
-
Beta-arrestin inhibits CAMKKbeta-dependent AMPK activation downstream of protease-activated-receptor-2.BMC Biochem. 2010 Sep 21;11:36. doi: 10.1186/1471-2091-11-36. BMC Biochem. 2010. PMID: 20858278 Free PMC article.
-
Molecular Mechanisms of Human Disease Mediated by Oncogenic and Primary Immunodeficiency Mutations in Class IA Phosphoinositide 3-Kinases.Front Immunol. 2018 Mar 19;9:575. doi: 10.3389/fimmu.2018.00575. eCollection 2018. Front Immunol. 2018. PMID: 29616047 Free PMC article. Review.
-
β-Arrestin-kinase scaffolds: turn them on or turn them off?Wiley Interdiscip Rev Syst Biol Med. 2013 Mar-Apr;5(2):231-41. doi: 10.1002/wsbm.1203. Epub 2013 Jan 14. Wiley Interdiscip Rev Syst Biol Med. 2013. PMID: 23319470 Review.
Cited by
-
Altered CXCR4 dynamics at the cell membrane impairs directed cell migration in WHIM syndrome patients.Proc Natl Acad Sci U S A. 2022 May 24;119(21):e2119483119. doi: 10.1073/pnas.2119483119. Epub 2022 May 19. Proc Natl Acad Sci U S A. 2022. PMID: 35588454 Free PMC article.
-
Proteinase-activated receptor 2 (PAR2) decreases apoptosis in colonic epithelial cells.J Biol Chem. 2014 Dec 5;289(49):34366-77. doi: 10.1074/jbc.M114.610485. Epub 2014 Oct 20. J Biol Chem. 2014. PMID: 25331954 Free PMC article.
-
Tissue factor-protease-activated receptor 2 signaling promotes diet-induced obesity and adipose inflammation.Nat Med. 2011 Oct 23;17(11):1490-7. doi: 10.1038/nm.2461. Nat Med. 2011. PMID: 22019885 Free PMC article.
-
Par2-mediated responses in inflammation and regeneration: choosing between repair and damage.Inflamm Regen. 2024 May 30;44(1):26. doi: 10.1186/s41232-024-00338-1. Inflamm Regen. 2024. PMID: 38816842 Free PMC article. Review.
-
The Diverse Roles of Arrestin Scaffolds in G Protein-Coupled Receptor Signaling.Pharmacol Rev. 2017 Jul;69(3):256-297. doi: 10.1124/pr.116.013367. Pharmacol Rev. 2017. PMID: 28626043 Free PMC article. Review.
References
-
- Zoudilova M., Kumar P., Ge L., Wang P., Bokoch G. M., DeFea K. A. β-arrestin-dependent regulation of the cofilin pathway downstream of protease-activated receptor-2. J. Biol. Chem. 2007;282:20634–20646. - PubMed
-
- Ge L., Ly Y., Hollenberg M., DeFea K. A β-arrestin-dependent scaffold is associated with prolonged MAPK activation in pseudopodia during protease-activated receptor-2-induced chemotaxis. J. Biol. Chem. 2003;278:34418–34426. - PubMed
-
- Ge L., Shenoy S. K., Lefkowitz R. J., DeFea K. A. Constitutive protease-activated-receptor-2 mediated migration of MDA MB-231 breast cancer cells requires both β-arrestin-1 and 2. J. Biol. Chem. 2004;279:55419–55424. - PubMed
-
- Jacob C., Yang P. C., Darmoul D., Amadesi S., Saito T., Cottrell G. S., Coelho A. M., Singh P., Grady E. F., Perdue M., Bunnett N. W. Mast cell tryptase controls paracellular permeability of the intestine. Role of protease-activated receptor 2 and β-arrestins. J. Biol. Chem. 2005;280:31936–31948. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous