

Lack of increases in methylation at three CpG-rich genomic loci in non-mitotic adult tissues during aging
- PMID: 17672908
- PMCID: PMC1950491
- DOI: 10.1186/1471-2350-8-50
Lack of increases in methylation at three CpG-rich genomic loci in non-mitotic adult tissues during aging
Abstract
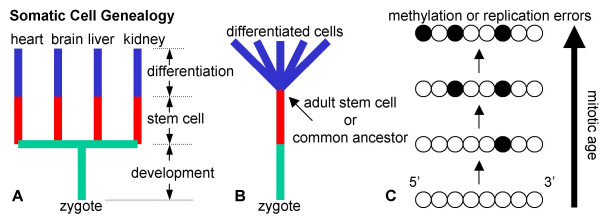
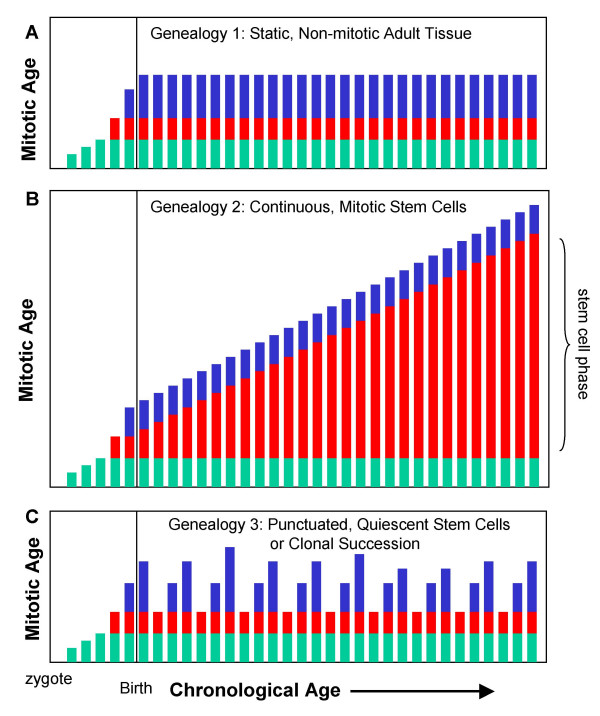
Background: Cell division occurs during normal human development and aging. Despite the likely importance of cell division to human pathology, it has been difficult to infer somatic cell mitotic ages (total numbers of divisions since the zygote) because direct counting of lifetime numbers of divisions is currently impractical. Here we attempt to infer relative mitotic ages with a molecular clock hypothesis. Somatic genomes may record their mitotic ages because greater numbers of replication errors should accumulate after greater numbers of divisions. Mitotic ages will vary between cell types if they divide at different times and rates.
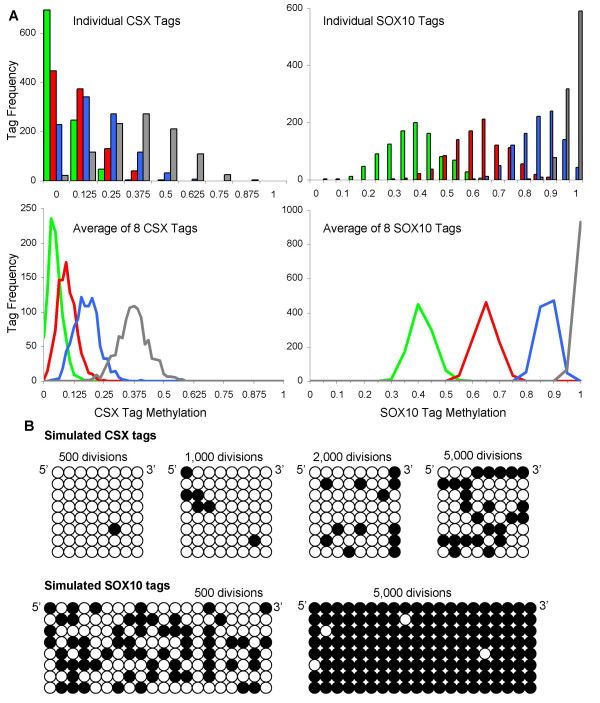
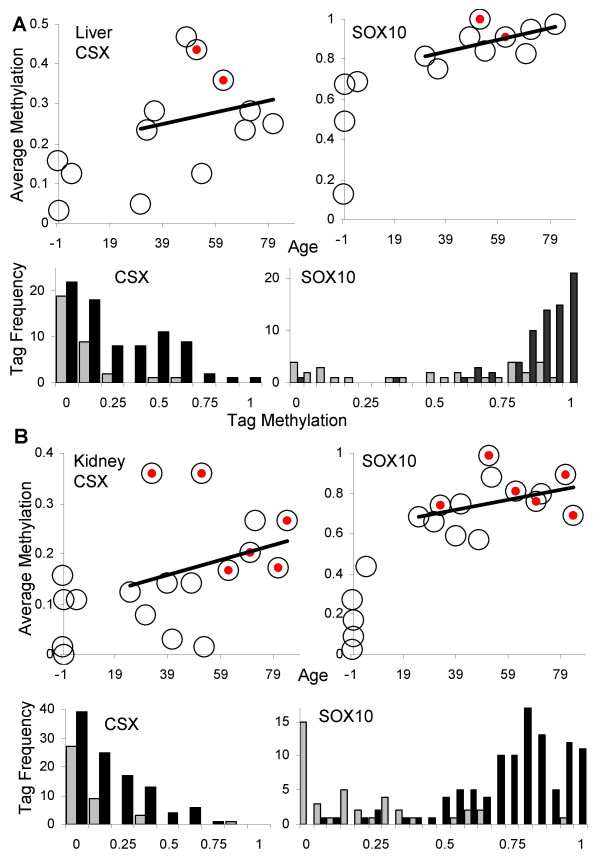
Methods: Age-related increases in DNA methylation at specific CpG sites (termed "epigenetic molecular clocks") have been previously observed in mitotic human epithelium like the intestines and endometrium. These CpG rich sequences or "tags" start unmethylated and potentially changes in methylation during development and aging represent replication errors. To help distinguish between mitotic versus time-associated changes, DNA methylation tag patterns at 8-20 CpGs within three different genes, two on autosomes and one on the X-chromosome were measured by bisulfite sequencing from heart, brain, kidney and liver of autopsies from 21 individuals of different ages.
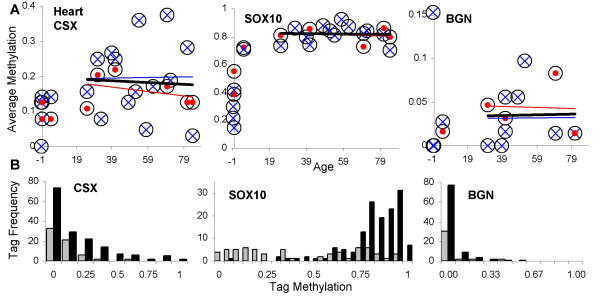
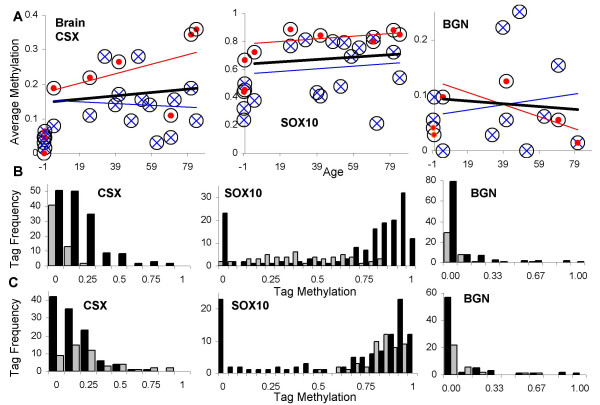
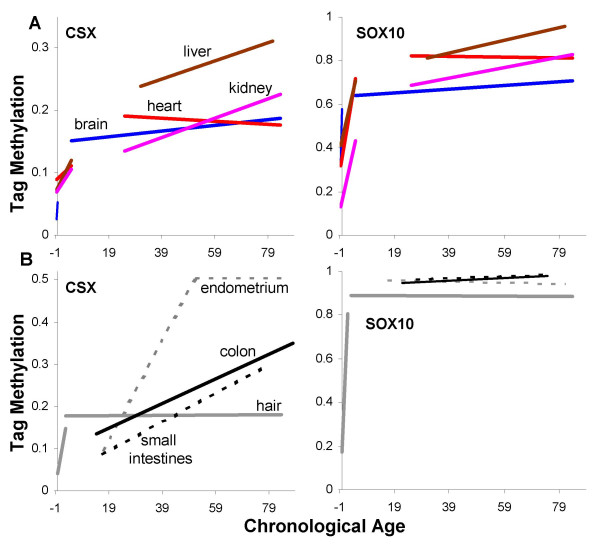
Results: Levels of DNA methylation were significantly greater in adult compared to fetal or newborn tissues for two of the three examined tags. Consistent with the relative absence of cell division in these adult tissues, there were no significant increases in tag methylation after infancy.
Conclusion: Many somatic methylation changes at certain CpG rich regions or tags appear to represent replication errors because this methylation increases with chronological age in mitotic epithelium but not in non-mitotic organs. Tag methylation accumulates differently in different tissues, consistent with their expected genealogies and mitotic ages. Although further studies are necessary, these results suggest numbers of divisions and ancestry are at least partially recorded by epigenetic replication errors within somatic cell genomes.
Figures

Similar articles
-
Inferring relative numbers of human leucocyte genome replications.Br J Haematol. 2008 Jun;141(6):862-71. doi: 10.1111/j.1365-2141.2008.07142.x. Epub 2008 Apr 10. Br J Haematol. 2008. PMID: 18410448
-
Age-related human small intestine methylation: evidence for stem cell niches.BMC Med. 2005 Jun 23;3:10. doi: 10.1186/1741-7015-3-10. BMC Med. 2005. PMID: 15975143 Free PMC article.
-
Older individuals appear to acquire mitotically older colorectal cancers.J Pathol. 2009 Mar;217(4):483-8. doi: 10.1002/path.2506. J Pathol. 2009. PMID: 19165870 Free PMC article.
-
Inferring human stem cell behaviour from epigenetic drift.J Pathol. 2009 Jan;217(2):199-205. doi: 10.1002/path.2461. J Pathol. 2009. PMID: 19031430 Free PMC article. Review.
-
Counting divisions in a human somatic cell tree: how, what and why?Cell Cycle. 2006 Mar;5(6):610-4. doi: 10.4161/cc.5.6.2570. Epub 2006 Mar 15. Cell Cycle. 2006. PMID: 16582617 Review.
Cited by
-
Passenger mutations as a marker of clonal cell lineages in emerging neoplasia.Semin Cancer Biol. 2010 Oct;20(5):294-303. doi: 10.1016/j.semcancer.2010.10.008. Epub 2010 Oct 15. Semin Cancer Biol. 2010. PMID: 20951806 Free PMC article. Review.
-
The relationship of DNA methylation with age, gender and genotype in twins and healthy controls.PLoS One. 2009 Aug 26;4(8):e6767. doi: 10.1371/journal.pone.0006767. PLoS One. 2009. PMID: 19774229 Free PMC article.
-
Promoter methylation and age-related downregulation of Klotho in rhesus monkey.Age (Dordr). 2012 Dec;34(6):1405-19. doi: 10.1007/s11357-011-9315-4. Epub 2011 Sep 16. Age (Dordr). 2012. PMID: 21922250 Free PMC article.
-
Aberrant intestinal stem cell lineage dynamics in Peutz-Jeghers syndrome and familial adenomatous polyposis consistent with protracted clonal evolution in the crypt.Gut. 2012 Jun;61(6):839-46. doi: 10.1136/gutjnl-2011-300622. Epub 2011 Sep 22. Gut. 2012. PMID: 21940722 Free PMC article.
-
Methylated eyes absent 4 (EYA4) gene promotor in non-neoplastic mucosa of ulcerative colitis patients with colorectal cancer: evidence for a field effect.Inflamm Bowel Dis. 2013 Sep;19(10):2079-83. doi: 10.1097/MIB.0b013e31829b3f4d. Inflamm Bowel Dis. 2013. PMID: 23867875 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
