

Chemogenomic approaches to rational drug design
- PMID: 17533416
- PMCID: PMC1978269
- DOI: 10.1038/sj.bjp.0707307
Chemogenomic approaches to rational drug design
Abstract
Paradigms in drug design and discovery are changing at a significant pace. Concomitant to the sequencing of over 180 several genomes, the high-throughput miniaturization of chemical synthesis and biological evaluation of a multiple compounds on gene/protein expression and function opens the way to global drug-discovery approaches, no more focused on a single target but on an entire family of related proteins or on a full metabolic pathway. Chemogenomics is this emerging research field aimed at systematically studying the biological effect of a wide array of small molecular-weight ligands on a wide array of macromolecular targets. Since the quantity of existing data (compounds, targets and assays) and of produced information (gene/protein expression levels and binding constants) are too large for manual manipulation, information technologies play a crucial role in planning, analysing and predicting chemogenomic data. The present review will focus on predictive in silico chemogenomic approaches to foster rational drug design and derive information from the simultaneous biological evaluation of multiple compounds on multiple targets. State-of-the-art methods for navigating in either ligand or target space will be presented and concrete drug design applications will be mentioned.
Figures

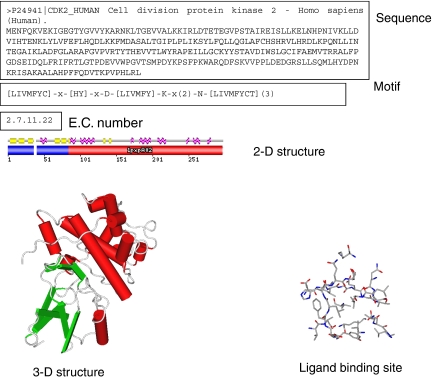


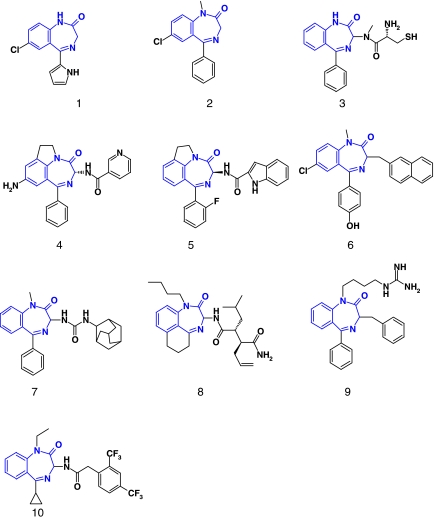
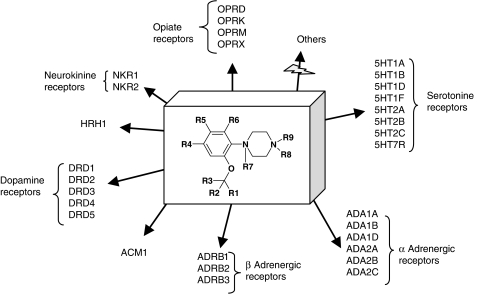

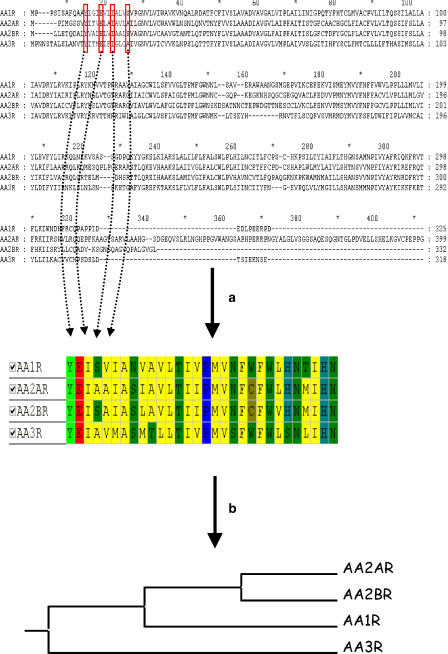



Comment in
-
Chemogenomic approaches to drug discovery: similar receptors bind similar ligands.Br J Pharmacol. 2007 Sep;152(1):5-7. doi: 10.1038/sj.bjp.0707308. Epub 2007 May 29. Br J Pharmacol. 2007. PMID: 17533415 Free PMC article.
Similar articles
-
Chemogenomic approaches to drug discovery: similar receptors bind similar ligands.Br J Pharmacol. 2007 Sep;152(1):5-7. doi: 10.1038/sj.bjp.0707308. Epub 2007 May 29. Br J Pharmacol. 2007. PMID: 17533415 Free PMC article.
-
In silico pharmacology for drug discovery: methods for virtual ligand screening and profiling.Br J Pharmacol. 2007 Sep;152(1):9-20. doi: 10.1038/sj.bjp.0707305. Epub 2007 Jun 4. Br J Pharmacol. 2007. PMID: 17549047 Free PMC article. Review.
-
Compound library design for target families.Methods Mol Biol. 2009;575:21-46. doi: 10.1007/978-1-60761-274-2_2. Methods Mol Biol. 2009. PMID: 19727610 Review.
-
In silico pharmacology for drug discovery: applications to targets and beyond.Br J Pharmacol. 2007 Sep;152(1):21-37. doi: 10.1038/sj.bjp.0707306. Epub 2007 Jun 4. Br J Pharmacol. 2007. PMID: 17549046 Free PMC article. Review.
-
Review on chemogenomics approach: interpreting antagonist activity of secreted frizzled-related protein 1 in glaucoma disease with in-silico docking.Curr Top Med Chem. 2012;12(16):1834-42. Curr Top Med Chem. 2012. PMID: 23030617 Review.
Cited by
-
Application of the quantum mechanical IEF/PCM-MST hydrophobic descriptors to selectivity in ligand binding.J Mol Model. 2016 Jun;22(6):136. doi: 10.1007/s00894-016-2991-3. Epub 2016 May 17. J Mol Model. 2016. PMID: 27188723
-
Chalcones as a basis for computer-aided drug design: innovative approaches to tackle malaria.Future Med Chem. 2019 Oct;11(20):2635-2646. doi: 10.4155/fmc-2018-0255. Epub 2019 Sep 26. Future Med Chem. 2019. PMID: 31556721 Free PMC article.
-
PharmMapper server: a web server for potential drug target identification using pharmacophore mapping approach.Nucleic Acids Res. 2010 Jul;38(Web Server issue):W609-14. doi: 10.1093/nar/gkq300. Epub 2010 Apr 29. Nucleic Acids Res. 2010. PMID: 20430828 Free PMC article.
-
Identification of Histamine H3 Receptor Ligands Using a New Crystal Structure Fragment-based Method.Sci Rep. 2017 Jul 6;7(1):4829. doi: 10.1038/s41598-017-05058-w. Sci Rep. 2017. PMID: 28684785 Free PMC article.
-
AlphaFold, allosteric, and orthosteric drug discovery: Ways forward.Drug Discov Today. 2023 Jun;28(6):103551. doi: 10.1016/j.drudis.2023.103551. Epub 2023 Mar 11. Drug Discov Today. 2023. PMID: 36907321 Free PMC article. Review.
References
-
- An J, Totrov M, Abagyan R. Pocketome via comprehensive identification and classification of ligand binding envelopes. Mol Cell Proteomics. 2005;4:752–761. - PubMed
-
- Artymiuk PJ, Poirrette AR, Grindley HM, Rice DW, Willett P. A graph-theoretic approach to the identification of three-dimensional patterns of amino acid side-chains in protein structures. J Mol Biol. 1994;243:327–344. - PubMed
-
- Bender A, Glen RC. Molecular similarity: a key technique in molecular informatics. Org Biomol Chem. 2004;2:3204–3218. - PubMed
-
- Bender A, Jenkins JL, Glick M, Deng Z, Nettles JH, Davies JW. ‘Bayes affinity fingerprints' improve retrieval rates in virtual screening and define orthogonal bioactivity space: when are multitarget drugs a feasible concept. J Chem In Model. 2006;46:2445–2456. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
