

Total syntheses, fragmentation studies, and antitumor/antiproliferative activities of FR901464 and its low picomolar analogue
- PMID: 17279752
- PMCID: PMC2530894
- DOI: 10.1021/ja067870m
Total syntheses, fragmentation studies, and antitumor/antiproliferative activities of FR901464 and its low picomolar analogue
Erratum in
- J Am Chem Soc. 2007 Jun 6;129(22):7206
Abstract
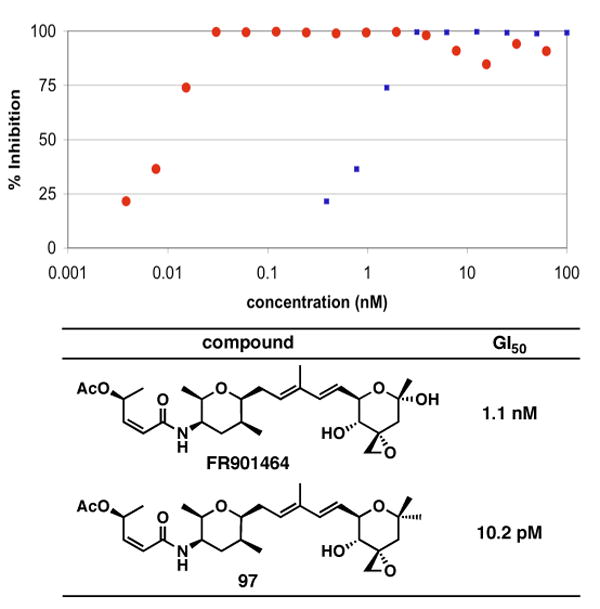
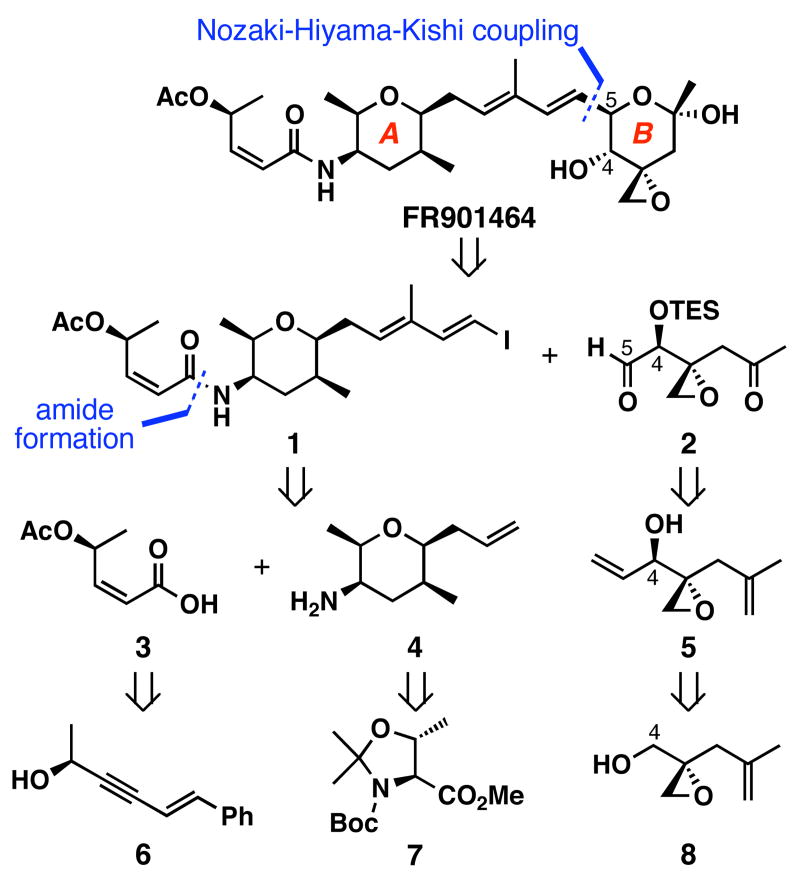
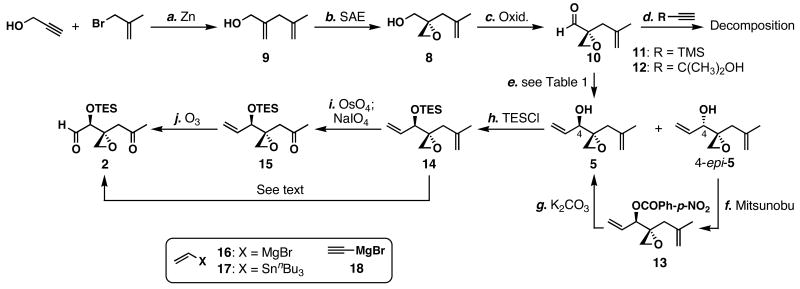
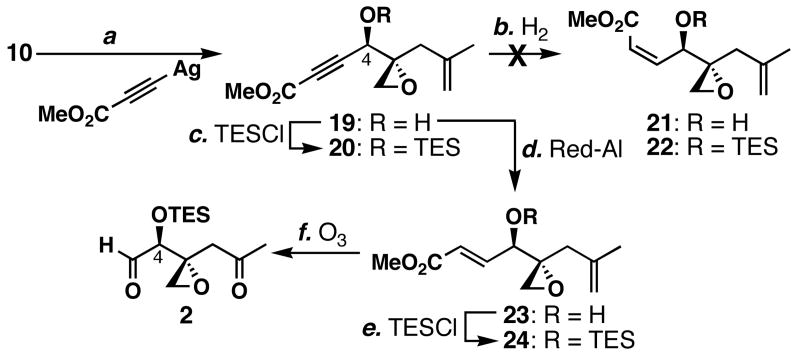
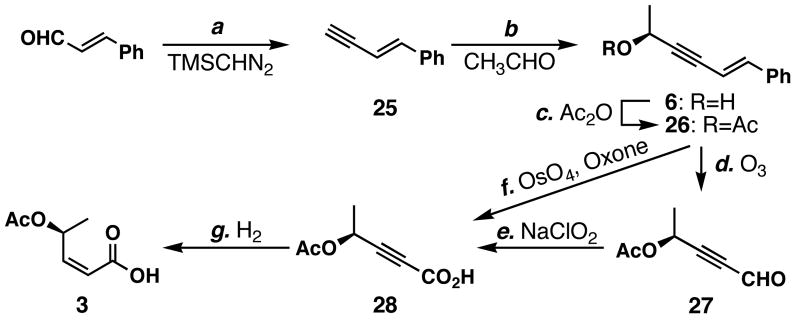
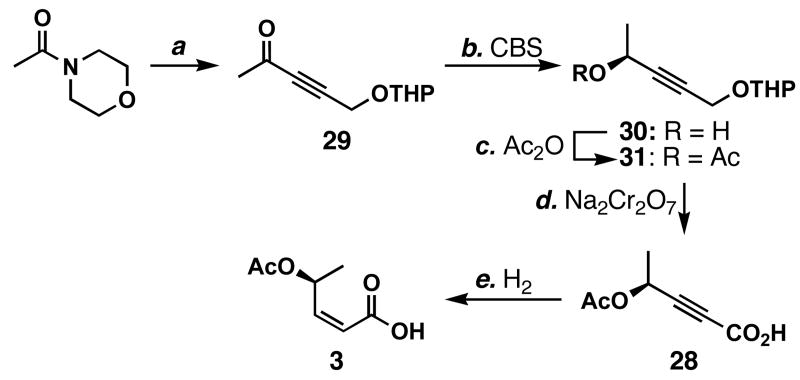
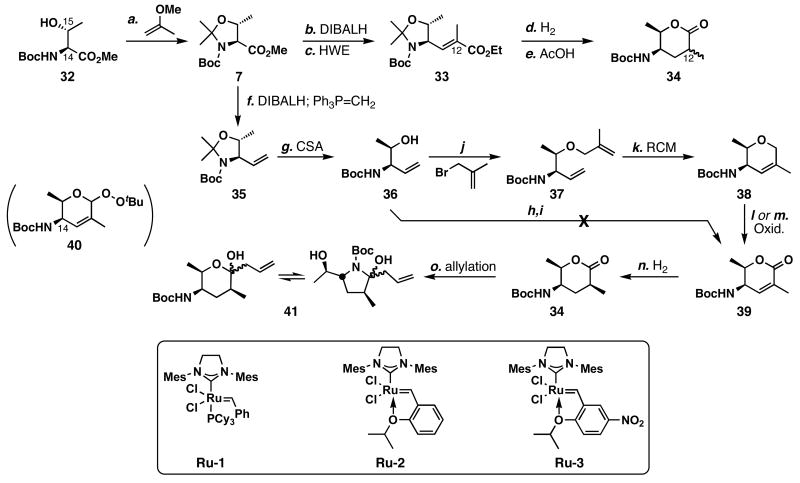
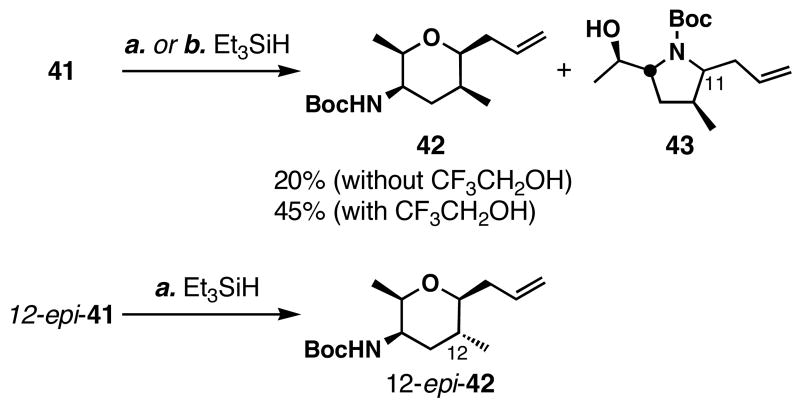
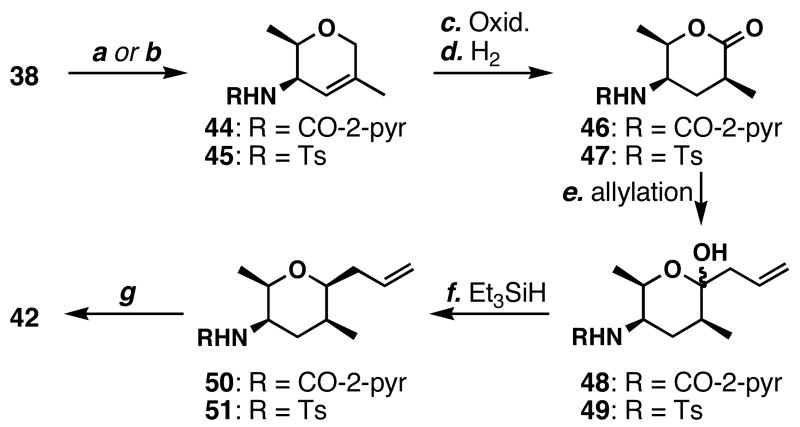
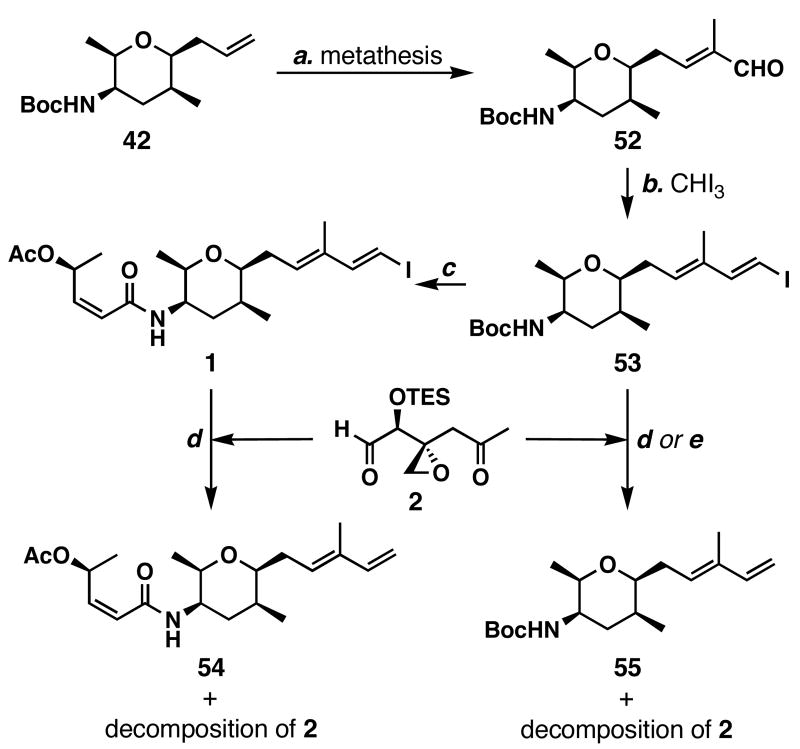
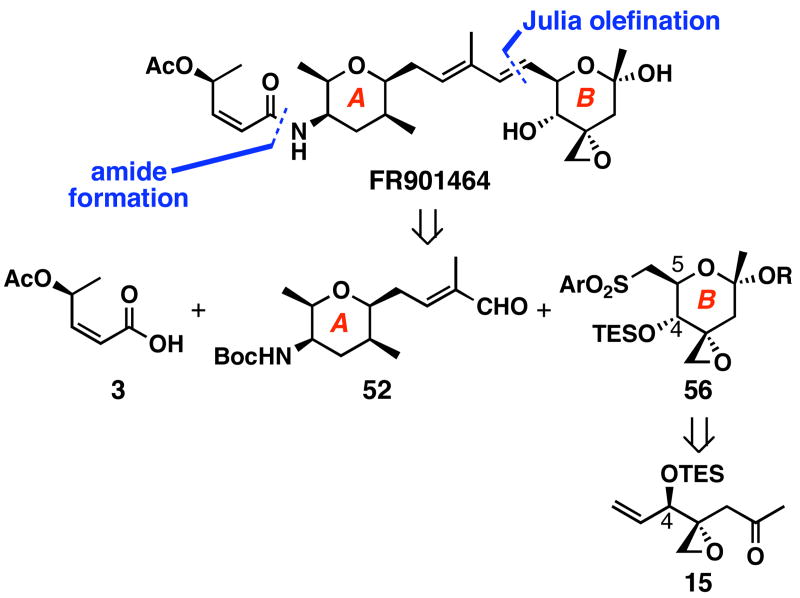
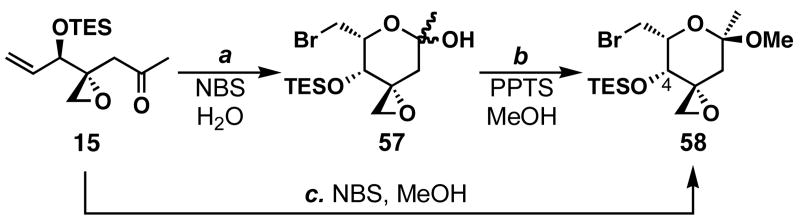
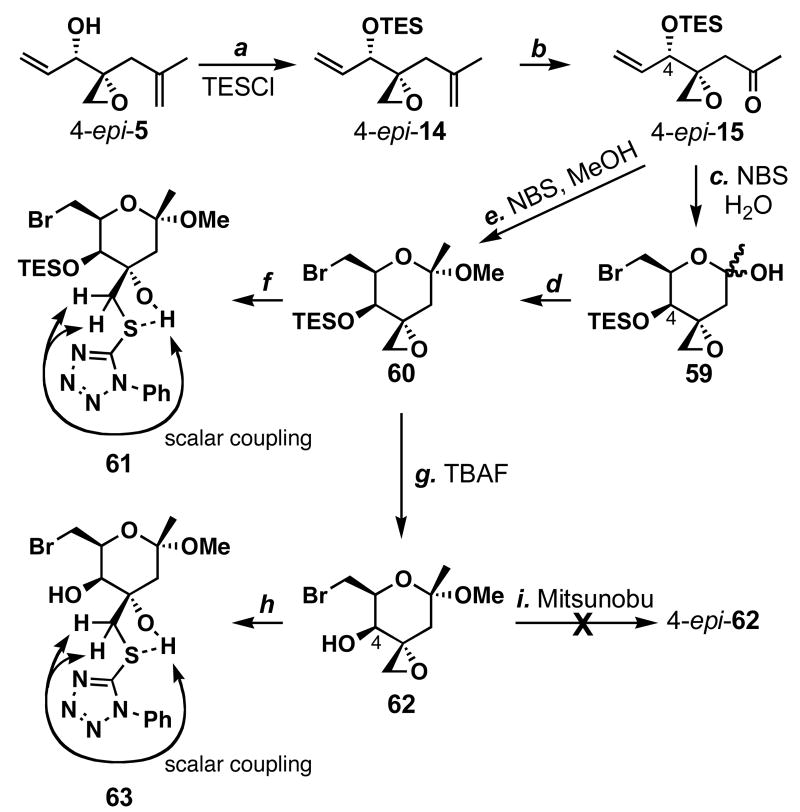
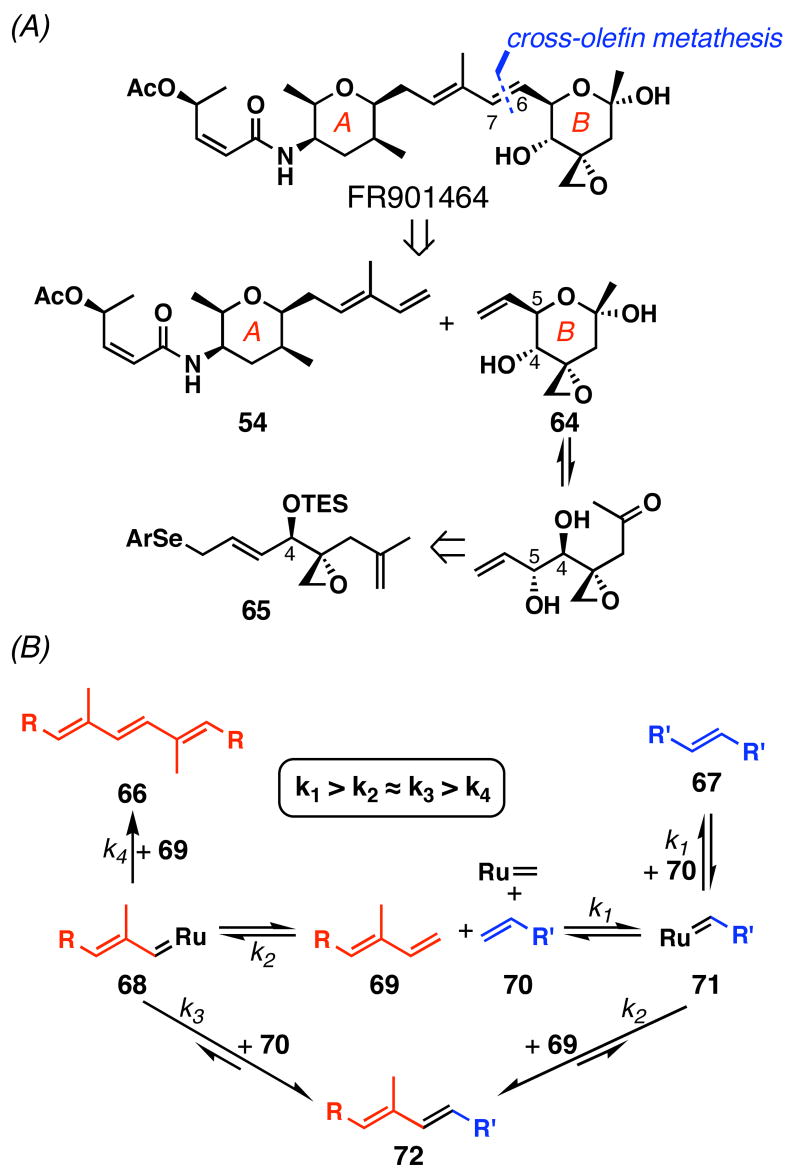
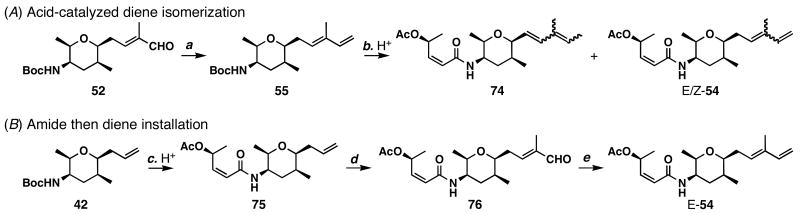
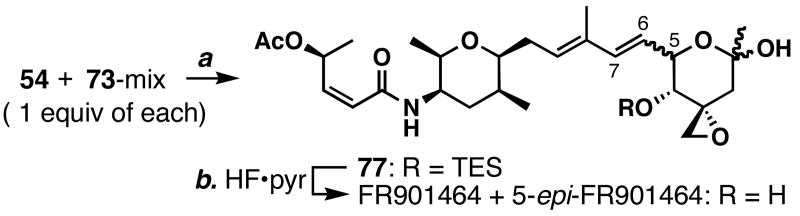
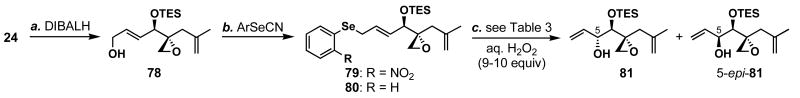
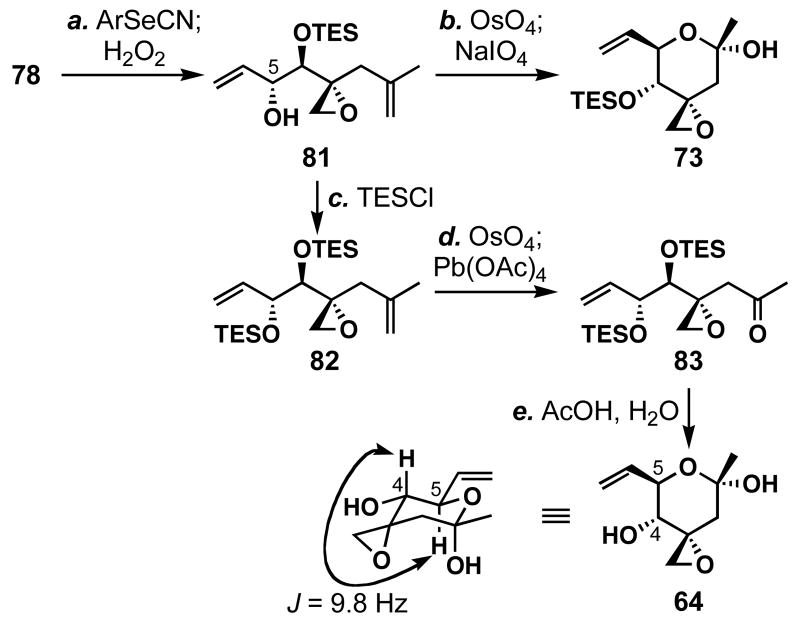
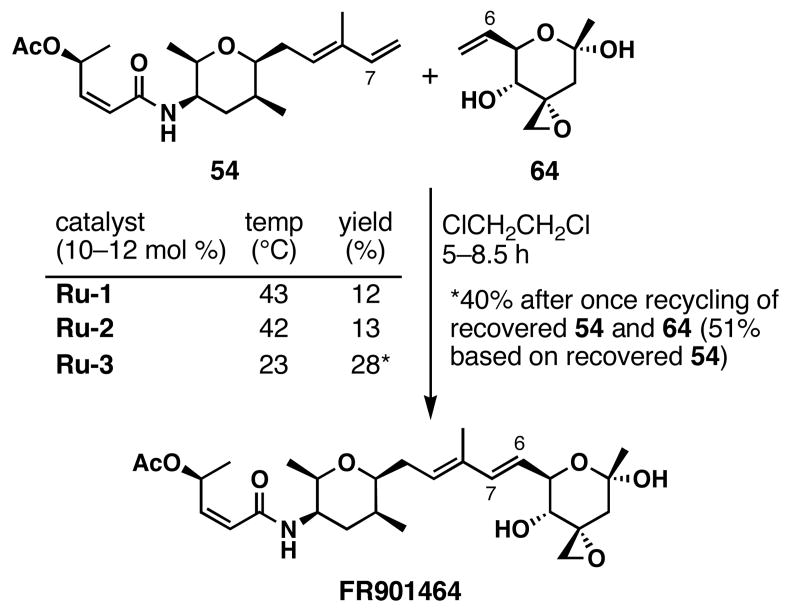
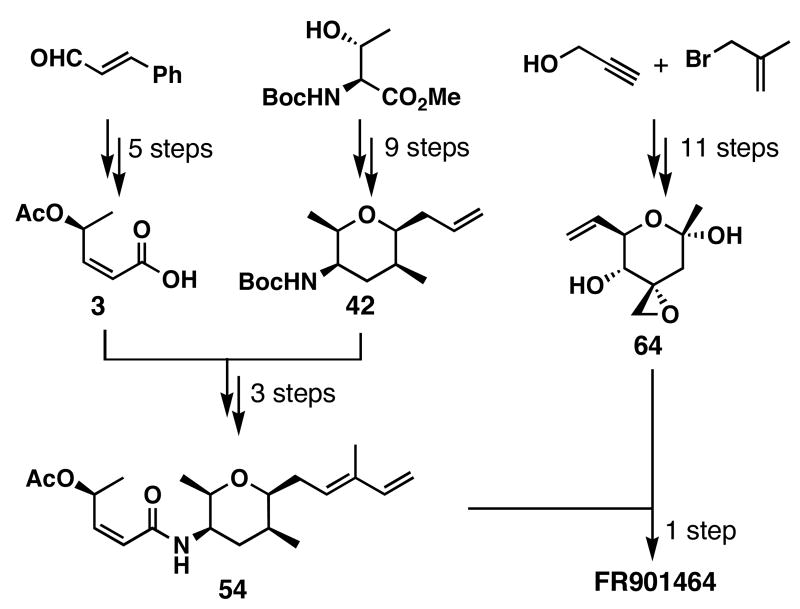
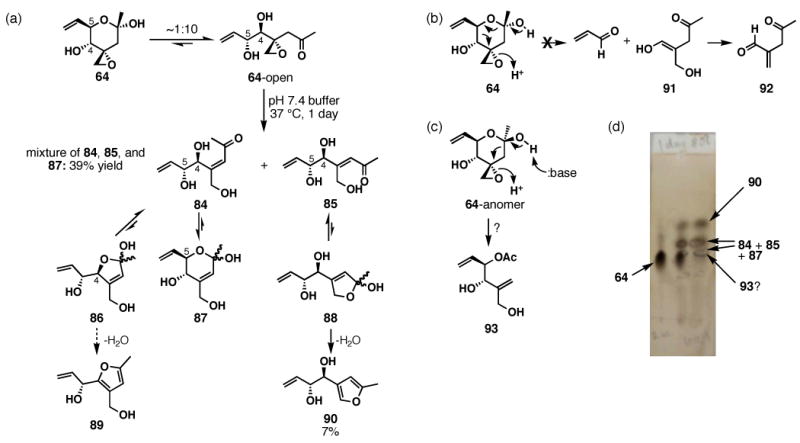
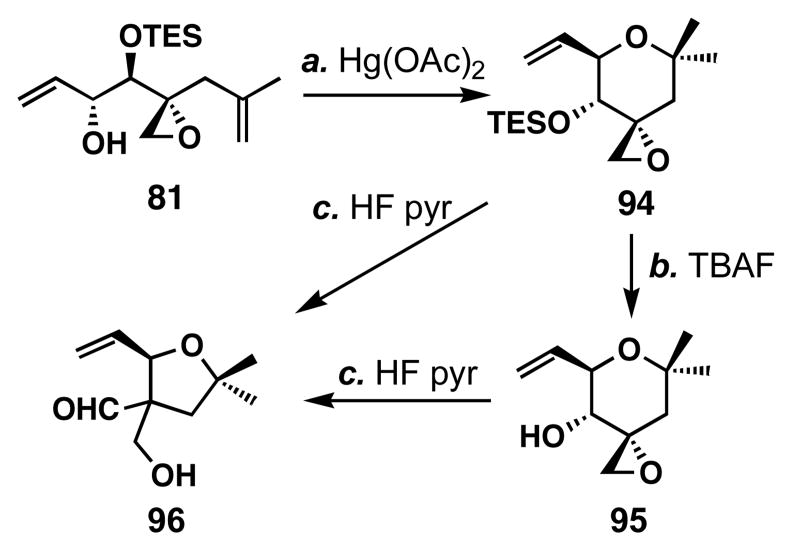
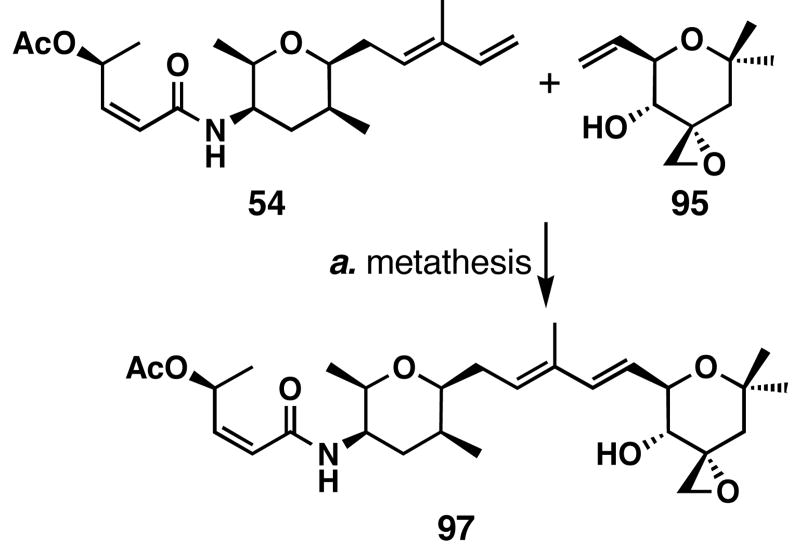
FR901464 is a potent anticancer natural product that lowers the mRNA levels of oncogenes and tumor suppressor genes. In this article, we report a convergent enantioselective synthesis of FR901464, which was accomplished in 13 linear steps. Central to the synthetic approach was the diene-ene cross olefin metathesis reaction to generate the C6-C7 olefin without the use of protecting groups as the final step. Additional key reactions include a Zr/Ag-promoted alkynylation to set the C4 stereocenter, a mild and chemoselective Red-Al reduction, a reagent-controlled stereoselective Mislow-Evans-type [2,3]-sigmatropic rearrangement to install the C5 stereocenter, a Carreira asymmetric alkynylation to generate the C4' stereocenter, and a highly efficient ring-closing metathesis-allylic oxidation sequence to form an unsaturated lactone. The decomposition pathways of FR901464's right fragment were studied under physiologically relevant conditions. Facile epoxide opening by beta-elimination gave two enones, one of which could undergo dehydration via its hemiketal to form a furan. To prevent this decomposition pathway, a right fragment was rationally designed and synthesized. This analogue was 12 times more stable than the right fragment of the natural product. Using this more stable right fragment analogue, an FR901464 analogue, meayamycin, was prepared in 13 linear steps. The inhibitions of human breast cancer MCF-7 cell proliferation by synthetic FR901464 and meayamycin were studied, and the GI50 values for these compounds were determined to be 1.1 nM and 10 pM, respectively. Thus, meayamycin is among the most potent anticancer small molecules that do not bind to either DNA or microtubule.
Figures
Similar articles
-
Total synthesis of FR901464, an antitumor agent that regulates the transcription of oncogenes and tumor suppressor genes.J Am Chem Soc. 2006 Mar 8;128(9):2792-3. doi: 10.1021/ja058216u. J Am Chem Soc. 2006. PMID: 16506745
-
Structural requirements for the antiproliferative activity of pre-mRNA splicing inhibitor FR901464.Chemistry. 2011 Jan 17;17(3):895-904. doi: 10.1002/chem.201002402. Epub 2010 Nov 19. Chemistry. 2011. PMID: 21226105 Free PMC article.
-
Synthesis and antiproliferative activity of a tetrahydrofuran analog of FR901464.Bioorg Med Chem Lett. 2024 May 15;104:129739. doi: 10.1016/j.bmcl.2024.129739. Epub 2024 Apr 8. Bioorg Med Chem Lett. 2024. PMID: 38599298
-
Cephalostatin analogues--synthesis and biological activity.Fortschr Chem Org Naturst. 2004;87:1-80. doi: 10.1007/978-3-7091-0581-8_1. Fortschr Chem Org Naturst. 2004. PMID: 15079895 Review.
-
SPIKET and COBRA compounds as novel tubulin modulators with potent anticancer activity.Curr Opin Investig Drugs. 2000 Oct;1(2):252-6. Curr Opin Investig Drugs. 2000. PMID: 11249582 Review.
Cited by
-
A piperidine chiron for the Veratrum alkaloids.J Org Chem. 2012 May 4;77(9):4235-41. doi: 10.1021/jo2026228. Epub 2012 Apr 13. J Org Chem. 2012. PMID: 22401662 Free PMC article.
-
mRNA transcript diversity creates new opportunities for pharmacological intervention.Mol Pharmacol. 2012 May;81(5):620-30. doi: 10.1124/mol.111.076604. Epub 2012 Feb 7. Mol Pharmacol. 2012. PMID: 22319206 Free PMC article. Review.
-
Enantioselective total syntheses of FR901464 and spliceostatin A and evaluation of splicing activity of key derivatives.J Org Chem. 2014 Jun 20;79(12):5697-709. doi: 10.1021/jo500800k. Epub 2014 May 30. J Org Chem. 2014. PMID: 24873648 Free PMC article.
-
Sterically Tuned N-Heterocyclic Carbene Ligands for the Efficient Formation of Hindered Products in Ru-Catalyzed Olefin Metathesis.ACS Catal. 2020 Oct 2;10(19):11394-11404. doi: 10.1021/acscatal.0c02770. Epub 2020 Sep 18. ACS Catal. 2020. PMID: 33123411 Free PMC article.
-
Modulating splicing with small molecular inhibitors of the spliceosome.Wiley Interdiscip Rev RNA. 2017 Mar;8(2):10.1002/wrna.1381. doi: 10.1002/wrna.1381. Epub 2016 Jul 21. Wiley Interdiscip Rev RNA. 2017. PMID: 27440103 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
