

MEGAN analysis of metagenomic data
- PMID: 17255551
- PMCID: PMC1800929
- DOI: 10.1101/gr.5969107
MEGAN analysis of metagenomic data
Abstract
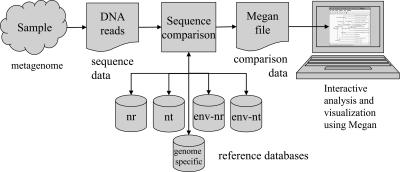
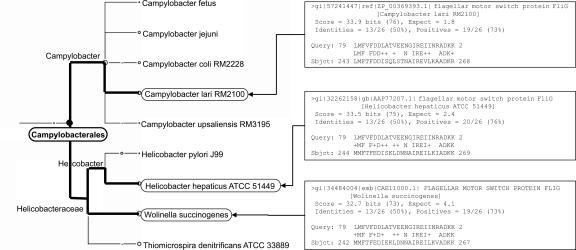
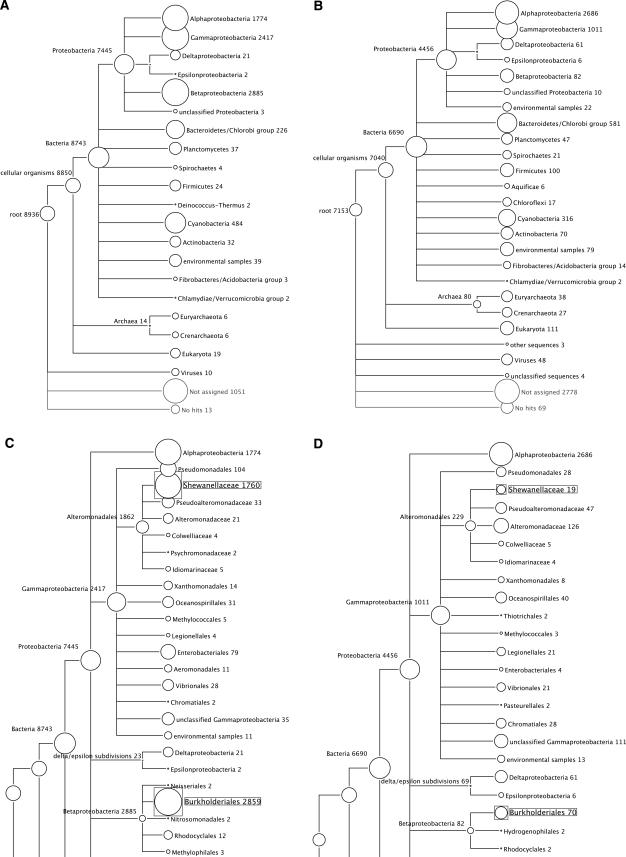
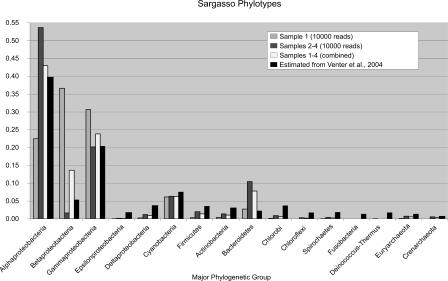
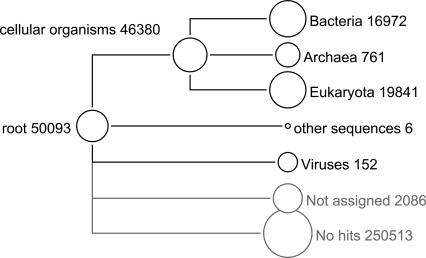
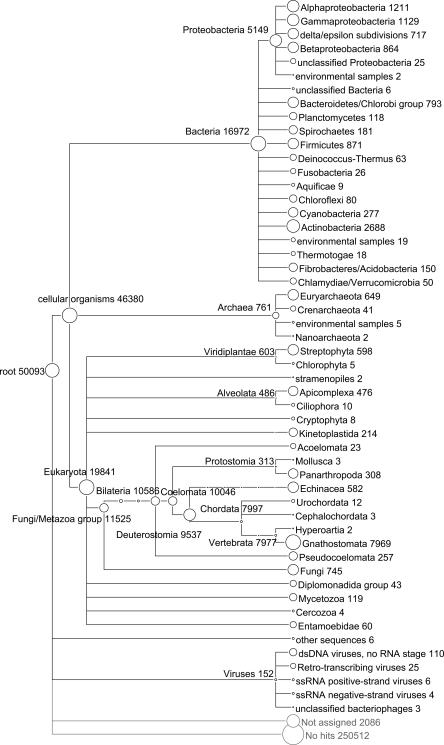
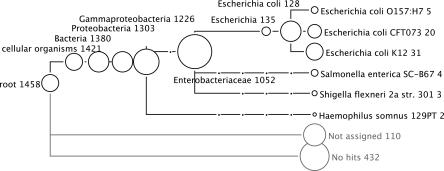
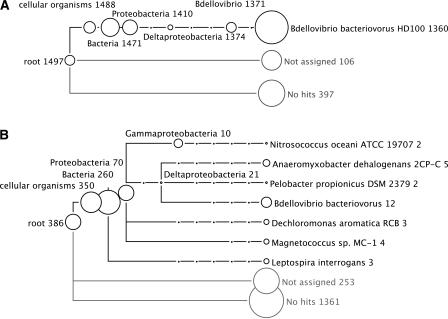
Metagenomics is the study of the genomic content of a sample of organisms obtained from a common habitat using targeted or random sequencing. Goals include understanding the extent and role of microbial diversity. The taxonomical content of such a sample is usually estimated by comparison against sequence databases of known sequences. Most published studies use the analysis of paired-end reads, complete sequences of environmental fosmid and BAC clones, or environmental assemblies. Emerging sequencing-by-synthesis technologies with very high throughput are paving the way to low-cost random "shotgun" approaches. This paper introduces MEGAN, a new computer program that allows laptop analysis of large metagenomic data sets. In a preprocessing step, the set of DNA sequences is compared against databases of known sequences using BLAST or another comparison tool. MEGAN is then used to compute and explore the taxonomical content of the data set, employing the NCBI taxonomy to summarize and order the results. A simple lowest common ancestor algorithm assigns reads to taxa such that the taxonomical level of the assigned taxon reflects the level of conservation of the sequence. The software allows large data sets to be dissected without the need for assembly or the targeting of specific phylogenetic markers. It provides graphical and statistical output for comparing different data sets. The approach is applied to several data sets, including the Sargasso Sea data set, a recently published metagenomic data set sampled from a mammoth bone, and several complete microbial genomes. Also, simulations that evaluate the performance of the approach for different read lengths are presented.
Figures

Similar articles
-
Use of simulated data sets to evaluate the fidelity of metagenomic processing methods.Nat Methods. 2007 Jun;4(6):495-500. doi: 10.1038/nmeth1043. Epub 2007 Apr 29. Nat Methods. 2007. PMID: 17468765
-
Metagenomics: read length matters.Appl Environ Microbiol. 2008 Mar;74(5):1453-63. doi: 10.1128/AEM.02181-07. Epub 2008 Jan 11. Appl Environ Microbiol. 2008. PMID: 18192407 Free PMC article.
-
Environmental genome shotgun sequencing of the Sargasso Sea.Science. 2004 Apr 2;304(5667):66-74. doi: 10.1126/science.1093857. Epub 2004 Mar 4. Science. 2004. PMID: 15001713
-
Get the most out of your metagenome: computational analysis of environmental sequence data.Curr Opin Microbiol. 2007 Oct;10(5):490-8. doi: 10.1016/j.mib.2007.09.001. Epub 2007 Oct 23. Curr Opin Microbiol. 2007. PMID: 17936679 Review.
-
Biodiversity assessment: state-of-the-art techniques in phylogenomics and species identification.Am J Bot. 2011 Mar;98(3):415-25. doi: 10.3732/ajb.1000296. Epub 2011 Feb 25. Am J Bot. 2011. PMID: 21613135 Review.
Cited by
-
Multifactorial diversity sustains microbial community stability.ISME J. 2013 Nov;7(11):2126-36. doi: 10.1038/ismej.2013.108. Epub 2013 Jul 4. ISME J. 2013. PMID: 23823494 Free PMC article.
-
Bayesian mixture analysis for metagenomic community profiling.Bioinformatics. 2015 Sep 15;31(18):2930-8. doi: 10.1093/bioinformatics/btv317. Epub 2015 May 21. Bioinformatics. 2015. PMID: 26002885 Free PMC article.
-
Analysis of composition-based metagenomic classification.BMC Genomics. 2012;13 Suppl 5(Suppl 5):S1. doi: 10.1186/1471-2164-13-S5-S1. Epub 2012 Oct 19. BMC Genomics. 2012. PMID: 23095761 Free PMC article.
-
Metagenomic datasets of air samples collected during episodes of severe smoke-haze in Malaysia.Data Brief. 2021 May 9;36:107124. doi: 10.1016/j.dib.2021.107124. eCollection 2021 Jun. Data Brief. 2021. PMID: 34095374 Free PMC article.
-
Metatranscriptomics Reveals the RNA Virome of Ixodes Persulcatus in the China-North Korea Border, 2017.Viruses. 2023 Dec 29;16(1):62. doi: 10.3390/v16010062. Viruses. 2023. PMID: 38257762 Free PMC article.
References
-
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J., Gish W., Miller W., Myers E.W., Lipman D.J., Miller W., Myers E.W., Lipman D.J., Myers E.W., Lipman D.J., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. - PubMed
-
- Béja O., Aravind L., Koonin E.V., Suzuki M.T., Hadd A., Nguyen L.P., Jovanovich S.B., Gates C.M., Feldman R.A., Spudich J.L., Aravind L., Koonin E.V., Suzuki M.T., Hadd A., Nguyen L.P., Jovanovich S.B., Gates C.M., Feldman R.A., Spudich J.L., Koonin E.V., Suzuki M.T., Hadd A., Nguyen L.P., Jovanovich S.B., Gates C.M., Feldman R.A., Spudich J.L., Suzuki M.T., Hadd A., Nguyen L.P., Jovanovich S.B., Gates C.M., Feldman R.A., Spudich J.L., Hadd A., Nguyen L.P., Jovanovich S.B., Gates C.M., Feldman R.A., Spudich J.L., Nguyen L.P., Jovanovich S.B., Gates C.M., Feldman R.A., Spudich J.L., Jovanovich S.B., Gates C.M., Feldman R.A., Spudich J.L., Gates C.M., Feldman R.A., Spudich J.L., Feldman R.A., Spudich J.L., Spudich J.L., et al. Bacterial rhodopsin: Evidence for a new type of phototrophy in the sea. Science. 2000;289:1902–1906. - PubMed
-
- Béja O., Spudich E.N., Spudich J.L., Leclerc M., DeLong E.F., Spudich E.N., Spudich J.L., Leclerc M., DeLong E.F., Spudich J.L., Leclerc M., DeLong E.F., Leclerc M., DeLong E.F., DeLong E.F. Proteorhodopsin phototrophy in the ocean. Nature. 2001;411:786–789. - PubMed
-
- Blattner F.R., Plunkett G., III, Bloch C.A., Perna N.T., Burland V., Riley M., Collado-Vides J., Glasner J.D., Rode C.K., Mayhew G.F., Plunkett G., III, Bloch C.A., Perna N.T., Burland V., Riley M., Collado-Vides J., Glasner J.D., Rode C.K., Mayhew G.F., Bloch C.A., Perna N.T., Burland V., Riley M., Collado-Vides J., Glasner J.D., Rode C.K., Mayhew G.F., Perna N.T., Burland V., Riley M., Collado-Vides J., Glasner J.D., Rode C.K., Mayhew G.F., Burland V., Riley M., Collado-Vides J., Glasner J.D., Rode C.K., Mayhew G.F., Riley M., Collado-Vides J., Glasner J.D., Rode C.K., Mayhew G.F., Collado-Vides J., Glasner J.D., Rode C.K., Mayhew G.F., Glasner J.D., Rode C.K., Mayhew G.F., Rode C.K., Mayhew G.F., Mayhew G.F., et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1474. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials