

Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signaling responses
- PMID: 17229704
- PMCID: PMC1866065
- DOI: 10.1128/JVI.02082-06
Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signaling responses
Abstract
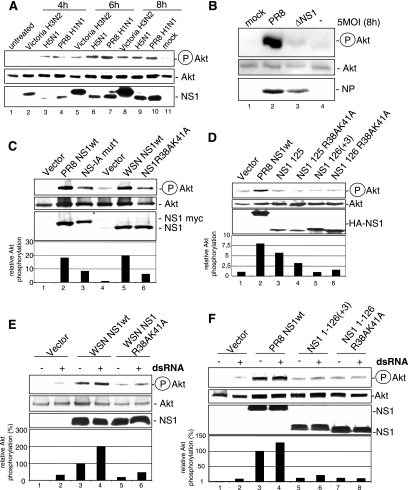
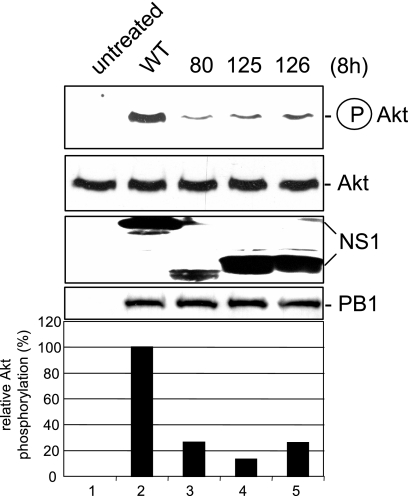
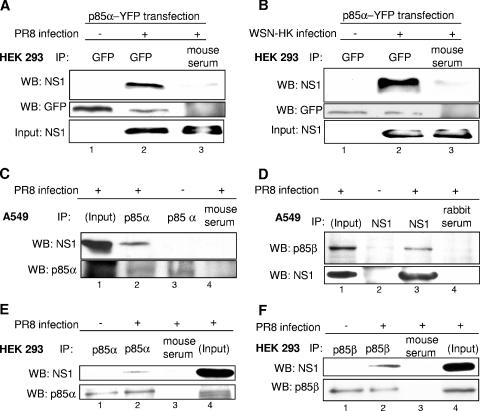
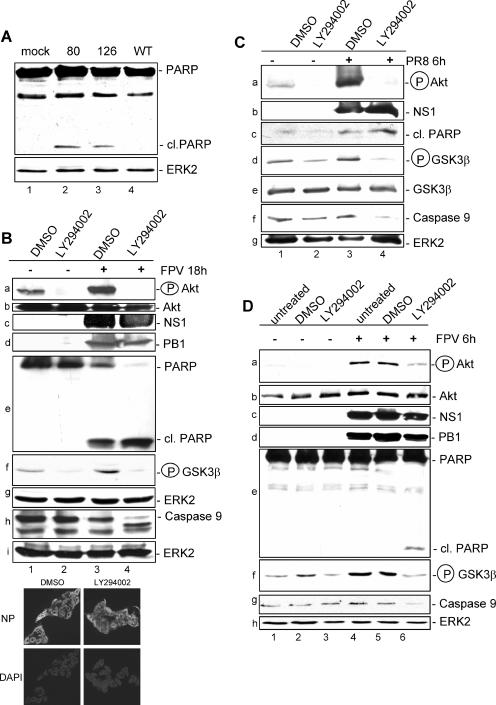
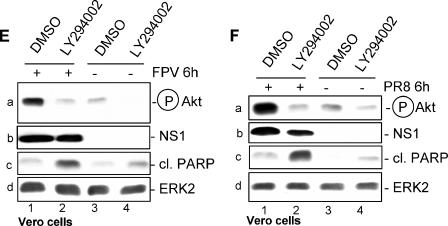
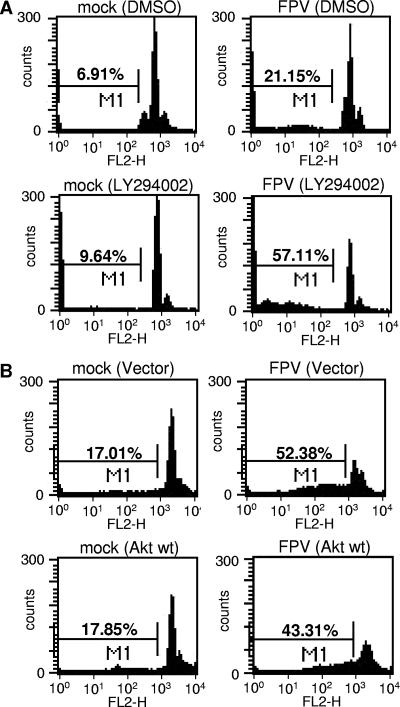
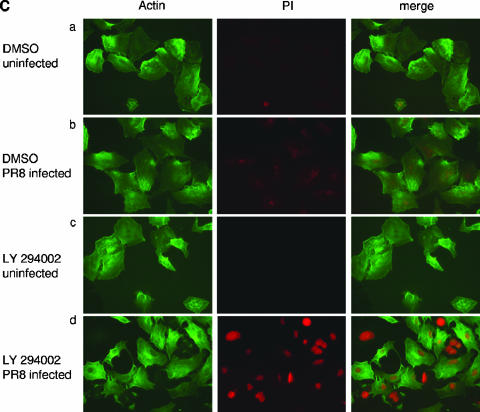
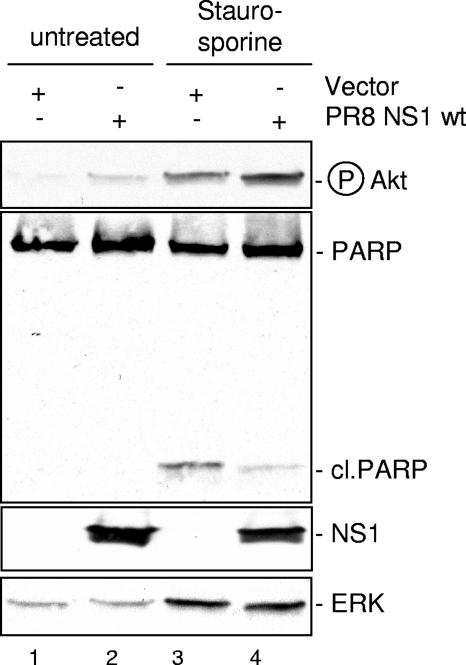
Recently we have shown that influenza A virus infection leads to activation of the phosphatidylinositol 3-kinase (PI3K)/Akt pathway and that this cellular reaction is dependent on the expression of the viral nonstructural protein 1 (NS1). These data also suggested that PI3K activation confers a virus-supporting activity at intermediate stages of the infection cycle. So far it is not known which process is regulated by the kinase that supports virus replication. It is well established that upon infection with influenza A virus, the expression of the viral NS1 keeps the induction of beta interferon and the apoptotic response within a tolerable limit. On a molecular basis, this activity of NS1 has been suggested to preclude the activation of cellular double-stranded RNA receptors as well as impaired modulation of mRNA processing. Here we present a novel mode of action of the NS1 protein to suppress apoptosis induction. NS1 binds to and activates PI3K, which results in the activation of the PI3K effector Akt. This leads to a subsequent inhibition of caspase 9 and glycogen synthase-kinase 3beta and limitation of the virus-induced cell death program. Thus, NS1 not only blocks but also activates signaling pathways to ensure efficient virus replication.
Figures
Similar articles
-
SH3 binding motif 1 in influenza A virus NS1 protein is essential for PI3K/Akt signaling pathway activation.J Virol. 2007 Dec;81(23):12730-9. doi: 10.1128/JVI.01427-07. Epub 2007 Sep 19. J Virol. 2007. PMID: 17881440 Free PMC article.
-
Activation of phosphatidylinositol 3-kinase signaling by the nonstructural NS1 protein is not conserved among type A and B influenza viruses.J Virol. 2007 Nov;81(21):12097-100. doi: 10.1128/JVI.01216-07. Epub 2007 Aug 22. J Virol. 2007. PMID: 17715214 Free PMC article.
-
Influenza A virus NS1 protein binds p85beta and activates phosphatidylinositol-3-kinase signaling.Proc Natl Acad Sci U S A. 2006 Sep 19;103(38):14194-9. doi: 10.1073/pnas.0606109103. Epub 2006 Sep 8. Proc Natl Acad Sci U S A. 2006. PMID: 16963558 Free PMC article.
-
A new player in a deadly game: influenza viruses and the PI3K/Akt signalling pathway.Cell Microbiol. 2009 Jun;11(6):863-71. doi: 10.1111/j.1462-5822.2009.01309.x. Epub 2009 Mar 12. Cell Microbiol. 2009. PMID: 19290913 Free PMC article. Review.
-
[Study progress of NS1 protein of influenza A virus].Wei Sheng Wu Xue Bao. 2007 Aug;47(4):729-33. Wei Sheng Wu Xue Bao. 2007. PMID: 17944383 Review. Chinese.
Cited by
-
In Vitro and In Vivo Antiviral Activity of Gingerenone A on Influenza A Virus Is Mediated by Targeting Janus Kinase 2.Viruses. 2020 Oct 8;12(10):1141. doi: 10.3390/v12101141. Viruses. 2020. PMID: 33050000 Free PMC article.
-
Analysis of miRNA expression in the trachea of Ri chicken infected with the highly pathogenic avian influenza H5N1 virus.J Vet Sci. 2023 Sep;24(5):e73. doi: 10.4142/jvs.23141. J Vet Sci. 2023. PMID: 38031652 Free PMC article.
-
Innate immune evasion strategies of influenza viruses.Future Microbiol. 2010 Jan;5(1):23-41. doi: 10.2217/fmb.09.108. Future Microbiol. 2010. PMID: 20020828 Free PMC article. Review.
-
Immunity to Influenza Infection in Humans.Cold Spring Harb Perspect Med. 2021 Mar 1;11(3):a038729. doi: 10.1101/cshperspect.a038729. Cold Spring Harb Perspect Med. 2021. PMID: 31871226 Free PMC article. Review.
-
Computational drug repurposing against SARS-CoV-2 reveals plasma membrane cholesterol depletion as key factor of antiviral drug activity.PLoS Comput Biol. 2022 Apr 11;18(4):e1010021. doi: 10.1371/journal.pcbi.1010021. eCollection 2022 Apr. PLoS Comput Biol. 2022. PMID: 35404937 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
