

Autoactivation of the Epstein-Barr virus oncogenic protein LMP1 during type II latency through opposite roles of the NF-kappaB and JNK signaling pathways
- PMID: 16840319
- PMCID: PMC1563735
- DOI: 10.1128/JVI.02052-05
Autoactivation of the Epstein-Barr virus oncogenic protein LMP1 during type II latency through opposite roles of the NF-kappaB and JNK signaling pathways
Abstract
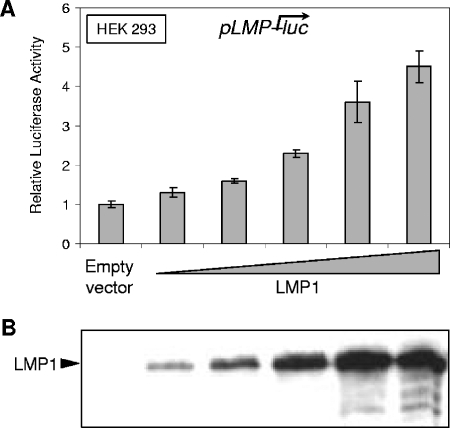
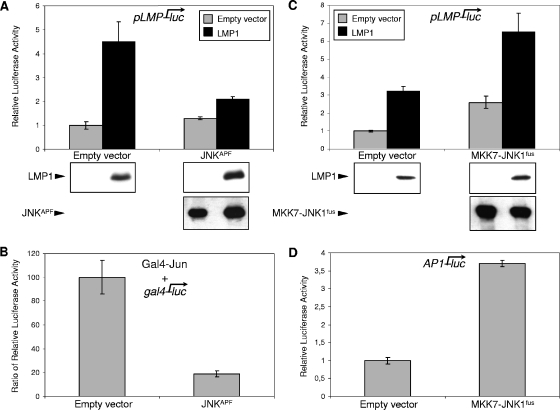
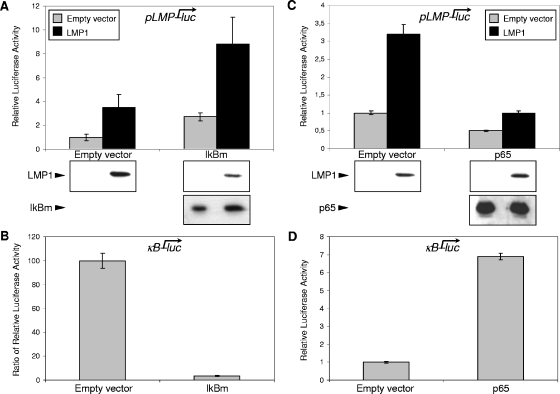
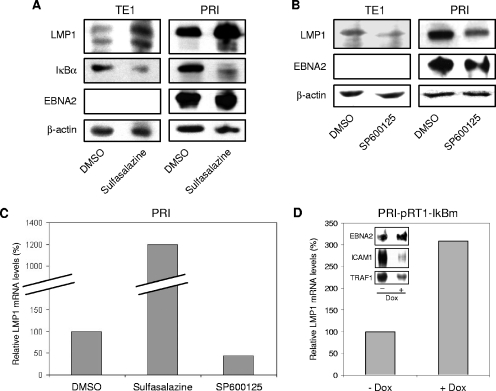
Epstein-Barr virus (EBV) is associated with several human malignancies where it expresses limited subsets of latent proteins. Of the latent proteins, latent membrane protein 1 (LMP1) is a potent transforming protein that constitutively induces multiple cell signaling pathways and contributes to EBV-associated oncogenesis. Regulation of LMP1 expression has been extensively described during the type III latency of EBV. Nevertheless, in the majority of EBV-associated tumors, the virus is commonly found to display a type II latency program in which it is still unknown which viral or cellular protein is really involved in maintaining LMP1 expression. Here, we demonstrate that LMP1 activates its own promoter pLMP1 through the JNK signaling pathway emerging from the TES2 domain. Our results also reveal that this activation is tightly controlled by LMP1, since pLMP1 is inhibited by LMP1-activated NF-kappaB signaling pathway. By using our physiological models of EBV-infected cells displaying type II latency as well as lymphoblastoid cell lines expressing a type III latency, we also demonstrate that this balanced autoregulation of LMP1 is shared by both latency programs. Finally, we show that this autoactivation is the most important mechanism to maintain LMP1 expression during the type II latency program of EBV.
Figures

Similar articles
-
A novel dominant-negative mutant form of Epstein-Barr virus latent membrane protein-1 (LMP1) selectively and differentially impairs LMP1 and TNF signaling pathways.Oncogene. 2004 Apr 8;23(15):2681-93. doi: 10.1038/sj.onc.1207432. Oncogene. 2004. PMID: 14767477
-
NF-κB Signaling Regulates Expression of Epstein-Barr Virus BART MicroRNAs and Long Noncoding RNAs in Nasopharyngeal Carcinoma.J Virol. 2016 Jun 24;90(14):6475-88. doi: 10.1128/JVI.00613-16. Print 2016 Jul 15. J Virol. 2016. PMID: 27147748 Free PMC article.
-
Maintenance of Epstein-Barr Virus Latent Status by a Novel Mechanism, Latent Membrane Protein 1-Induced Interleukin-32, via the Protein Kinase Cδ Pathway.J Virol. 2015 Jun;89(11):5968-80. doi: 10.1128/JVI.00168-15. Epub 2015 Mar 25. J Virol. 2015. PMID: 25810549 Free PMC article.
-
The Latent Membrane Protein 1 (LMP1).Curr Top Microbiol Immunol. 2015;391:119-49. doi: 10.1007/978-3-319-22834-1_4. Curr Top Microbiol Immunol. 2015. PMID: 26428373 Review.
-
The role of Epstein-Barr virus in neoplastic transformation.Oncology. 2001;60(4):289-302. doi: 10.1159/000058523. Oncology. 2001. PMID: 11408795 Review.
Cited by
-
LMP1-induced cell death may contribute to the emergency of its oncogenic property.PLoS One. 2013 Apr 23;8(4):e60743. doi: 10.1371/journal.pone.0060743. Print 2013. PLoS One. 2013. PMID: 23637765 Free PMC article.
-
Epstein-Barr Virus-associated lymphoproliferative disorders: experimental and clinical developments.Int J Clin Exp Med. 2015 Sep 15;8(9):14656-71. eCollection 2015. Int J Clin Exp Med. 2015. PMID: 26628948 Free PMC article. Review.
-
Resveratrol prevents EBV transformation and inhibits the outgrowth of EBV-immortalized human B cells.PLoS One. 2012;7(12):e51306. doi: 10.1371/journal.pone.0051306. Epub 2012 Dec 10. PLoS One. 2012. PMID: 23251493 Free PMC article.
-
Burkitt's lymphoma: the Rosetta Stone deciphering Epstein-Barr virus biology.Semin Cancer Biol. 2009 Dec;19(6):377-88. doi: 10.1016/j.semcancer.2009.07.004. Epub 2009 Jul 18. Semin Cancer Biol. 2009. PMID: 19619657 Free PMC article. Review.
-
Modulation of alternative splicing during early infection of human primary B lymphocytes with Epstein-Barr virus (EBV): a novel function for the viral EBNA-LP protein.Nucleic Acids Res. 2021 Oct 11;49(18):10657-10676. doi: 10.1093/nar/gkab787. Nucleic Acids Res. 2021. PMID: 34530456 Free PMC article.
References
-
- Adamson, A. L., D. Darr, E. Holley-Guthrie, R. A. Johnson, A. Mauser, J. Swenson, and S. Kenney. 2000. Epstein-Barr virus immediate-early proteins BZLF1 and BRLF1 activate the ATF2 transcription factor by increasing the levels of phosphorylated p38 and c-Jun N-terminal kinases. J. Virol. 74:1224-1233. - PMC - PubMed
-
- Adriaenssens, E., A. Mougel, G. Goormachtigh, E. Loing, V. Fafeur, C. Auriault, and J. Coll. 2004. A novel dominant-negative mutant form of Epstein-Barr virus latent membrane protein-1 (LMP1) selectively and differentially impairs LMP1 and TNF signaling pathways. Oncogene 23:2681-2693. - PubMed
-
- Bennett, B. L., D. T. Sasaki, B. W. Murray, E. C. O'Leary, S. T. Sakata, W. Xu, J. C. Leisten, A. Motiwala, S. Pierce, Y. Satoh, S. S. Bhagwat, A. M. Manning, and D. W. Anderson. 2001. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 98:13681-13686. - PMC - PubMed
-
- Bornkamm, G. W., C. Berens, C. Kuklik-Roos, J. M. Bechet, G. Laux, J. Bachl, M. Korndoerfer, M. Schlee, M. Hölzel, A. Malamoussi, R. D. Chapman, F. Nimmerjahn, J. Mautner, W. Hillen, H. Bujard, and J. Feuillard. 7 September 2005, posting date. Stringent doxycycline-dependent control of gene activities using an episomal one-vector system. Nucleic Acids Res. 33:e137. [Online.] doi:10.1093/nar/gni137. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
