

ST6Gal-I restrains CD22-dependent antigen receptor endocytosis and Shp-1 recruitment in normal and pathogenic immune signaling
- PMID: 16782884
- PMCID: PMC1489171
- DOI: 10.1128/MCB.00308-06
ST6Gal-I restrains CD22-dependent antigen receptor endocytosis and Shp-1 recruitment in normal and pathogenic immune signaling
Abstract
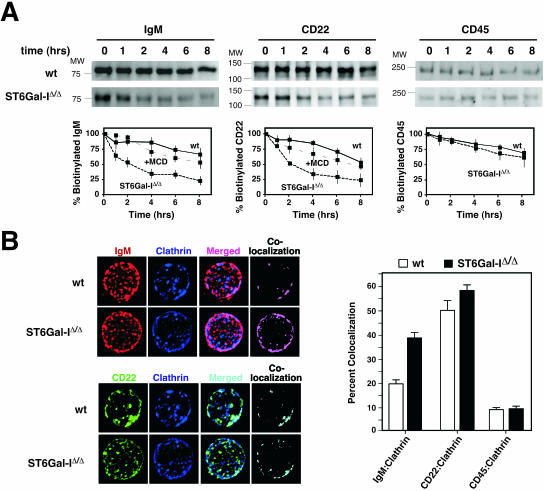
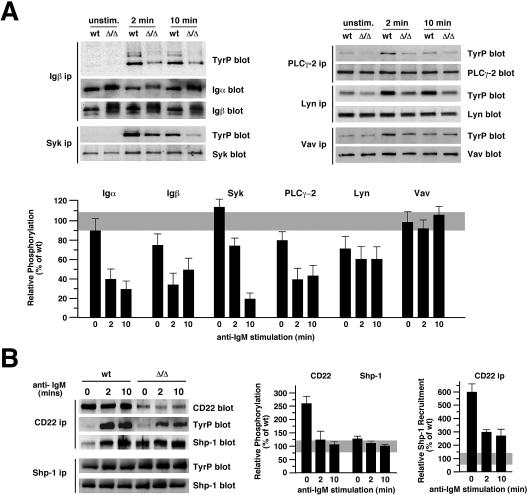
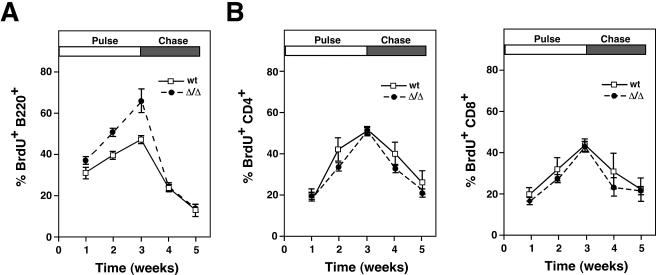
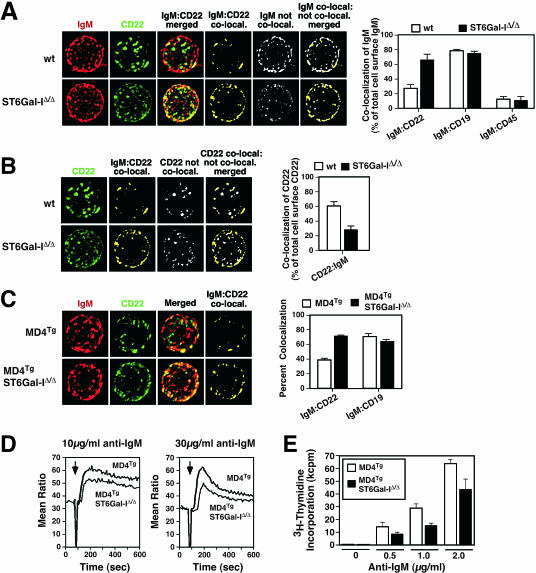
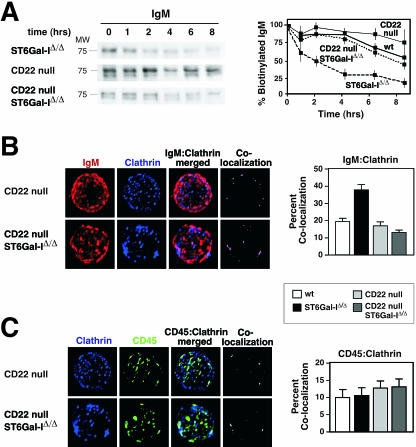
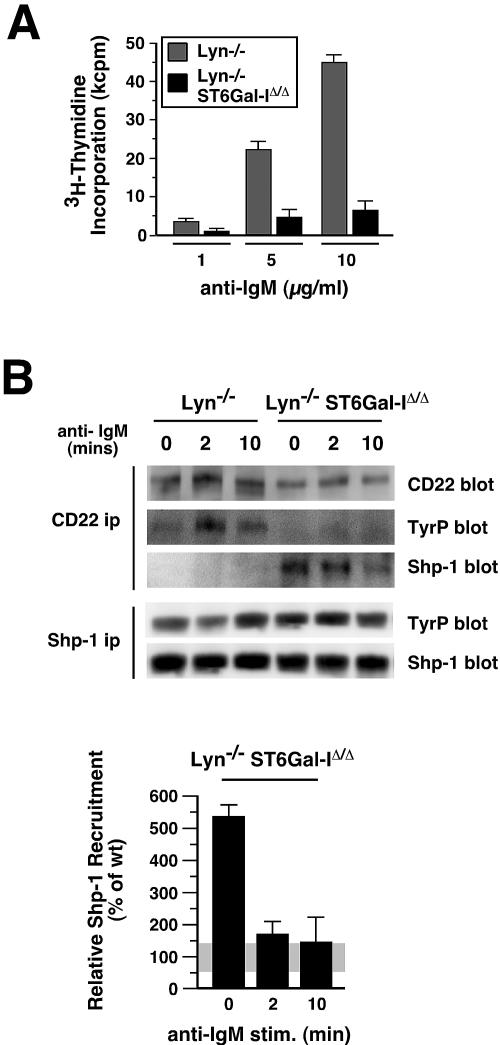
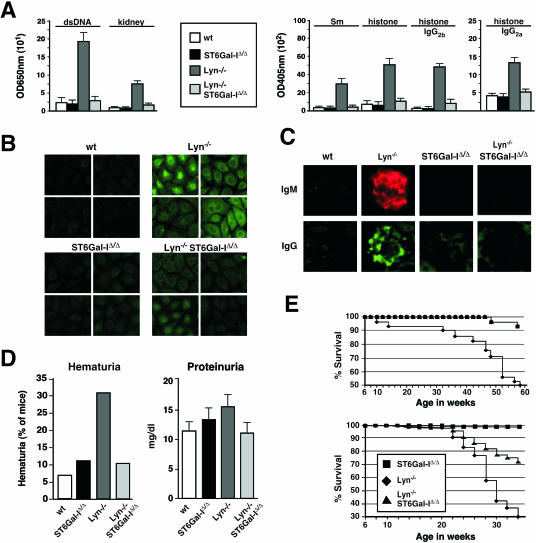
The ST6Gal-I sialyltransferase produces Siglec ligands for the B-cell-specific CD22 lectin and sustains humoral immune responses. Using multiple experimental approaches to elucidate the mechanisms involved, we report that ST6Gal-I deficiency induces immunoglobulin M (IgM) antigen receptor endocytosis in the absence of immune stimulation. This coincides with increased antigen receptor colocalization with CD22 in both clathrin-deficient and clathrin-enriched membrane microdomains concurrent with diminished tyrosine phosphorylation of Igalpha/beta, Syk, and phospholipase C-gamma2 upon immune activation. Codeficiency with CD22 restores IgM antigen receptor half-life at the cell surface in addition to reversing alterations in membrane trafficking and immune signaling. Diminished immune responses due to ST6Gal-I deficiency further correlate with constitutive recruitment of Shp-1 to CD22 in unstimulated B cells independent of Lyn tyrosine kinase activity and prevent autoimmune disease pathogenesis in the Lyn-deficient model of systemic lupus erythematosus, resulting in a significant extension of life span. Protein glycosylation by ST6Gal-I restricts access of antigen receptors and Shp-1 to CD22 and operates by a CD22-dependent mechanism that decreases the basal rate of IgM antigen receptor endocytosis in altering the threshold of B-cell immune activation.
Figures
Similar articles
-
Ablation of CD22 in ligand-deficient mice restores B cell receptor signaling.Nat Immunol. 2006 Feb;7(2):199-206. doi: 10.1038/ni1283. Epub 2005 Dec 20. Nat Immunol. 2006. PMID: 16369536
-
Polygenic autoimmune traits: Lyn, CD22, and SHP-1 are limiting elements of a biochemical pathway regulating BCR signaling and selection.Immunity. 1998 Apr;8(4):497-508. doi: 10.1016/s1074-7613(00)80554-3. Immunity. 1998. PMID: 9586639
-
Expression of B cell receptor-associated signaling molecules in human lupus.Autoimmunity. 2001 May;33(3):213-24. doi: 10.3109/08916930109008048. Autoimmunity. 2001. PMID: 11683380
-
Signaling mutations and autoimmunity.Curr Dir Autoimmun. 2003;6:61-88. doi: 10.1159/000066856. Curr Dir Autoimmun. 2003. PMID: 12408047 Review.
-
CD22 and Siglec-G regulate inhibition of B-cell signaling by sialic acid ligand binding and control B-cell tolerance.Glycobiology. 2014 Sep;24(9):807-17. doi: 10.1093/glycob/cwu066. Epub 2014 Jul 6. Glycobiology. 2014. PMID: 25002414 Review.
Cited by
-
Alpha2,6-sialic acid on platelet endothelial cell adhesion molecule (PECAM) regulates its homophilic interactions and downstream antiapoptotic signaling.J Biol Chem. 2010 Feb 26;285(9):6515-21. doi: 10.1074/jbc.M109.073106. Epub 2010 Jan 4. J Biol Chem. 2010. PMID: 20048157 Free PMC article.
-
CD22: an inhibitory enigma.Immunology. 2008 Mar;123(3):314-25. doi: 10.1111/j.1365-2567.2007.02752.x. Epub 2007 Dec 7. Immunology. 2008. PMID: 18067554 Free PMC article. Review.
-
CD22 is a recycling receptor that can shuttle cargo between the cell surface and endosomal compartments of B cells.J Immunol. 2011 Feb 1;186(3):1554-63. doi: 10.4049/jimmunol.1003005. Epub 2010 Dec 22. J Immunol. 2011. PMID: 21178016 Free PMC article.
-
Distinct endocytic mechanisms of CD22 (Siglec-2) and Siglec-F reflect roles in cell signaling and innate immunity.Mol Cell Biol. 2007 Aug;27(16):5699-710. doi: 10.1128/MCB.00383-07. Epub 2007 Jun 11. Mol Cell Biol. 2007. PMID: 17562860 Free PMC article.
-
The Glycoscience of Immunity.Trends Immunol. 2018 Jul;39(7):523-535. doi: 10.1016/j.it.2018.04.004. Epub 2018 May 11. Trends Immunol. 2018. PMID: 29759949 Free PMC article. Review.
References
-
- Angata, K., J. M. Long, O. Bukalo, W. Lee, A. Ditayev, A. Wynshaw-Boris, M. Schachner, M. Fukuda, and J. D. Marth. 2004. Sialyltransferase ST8Sia-II assembles a subset of polysialic acid that directs hippocampal axonal targeting and promotes fear behavior. J. Biol. Chem. 279:32603-32613. - PubMed
-
- Baum, L. 2002. Developing a taste for sweets. Immunity 16:5-8. - PubMed
-
- Chan, V. W. F., F. Meng, P. Soriano, A. L. DeFranco, and C. A. Lowell. 1997. Characterization of the B lymphocyte populations in Lyn-deficient mice and the role of Lyn in signal transduction and down-regulation. Immunity 7:69-81. - PubMed
-
- Collins, B. E., O. Blixt, N. V. Bovin, C. P. Danzer, D. Chui, J. D. Marth, L. Nitschke, and J. C. Paulson. 2002. Constitutively unmasked CD22 on B cells of ST6Gal I knockout mice: novel sialoside probe for murine CD22. Glycobiology 12:563-571. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous