

Comprehensive curation and analysis of global interaction networks in Saccharomyces cerevisiae
- PMID: 16762047
- PMCID: PMC1561585
- DOI: 10.1186/jbiol36
Comprehensive curation and analysis of global interaction networks in Saccharomyces cerevisiae
Abstract
Background: The study of complex biological networks and prediction of gene function has been enabled by high-throughput (HTP) methods for detection of genetic and protein interactions. Sparse coverage in HTP datasets may, however, distort network properties and confound predictions. Although a vast number of well substantiated interactions are recorded in the scientific literature, these data have not yet been distilled into networks that enable system-level inference.
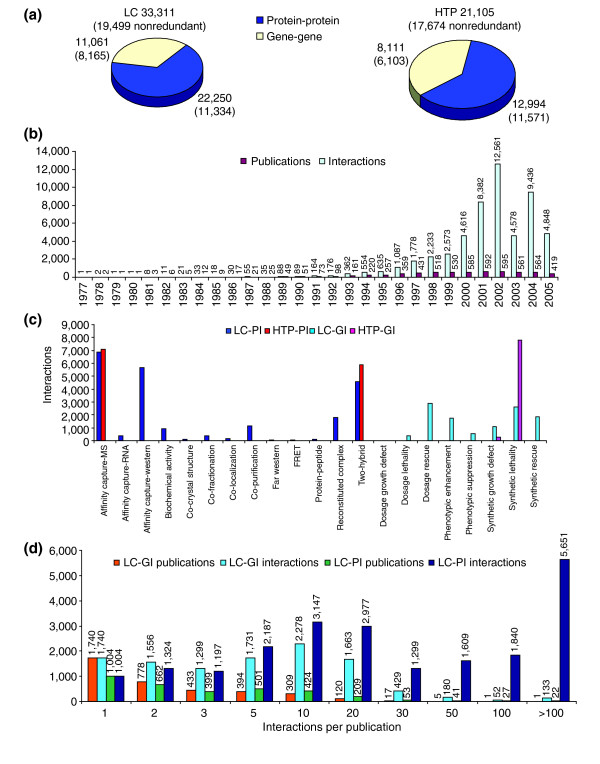
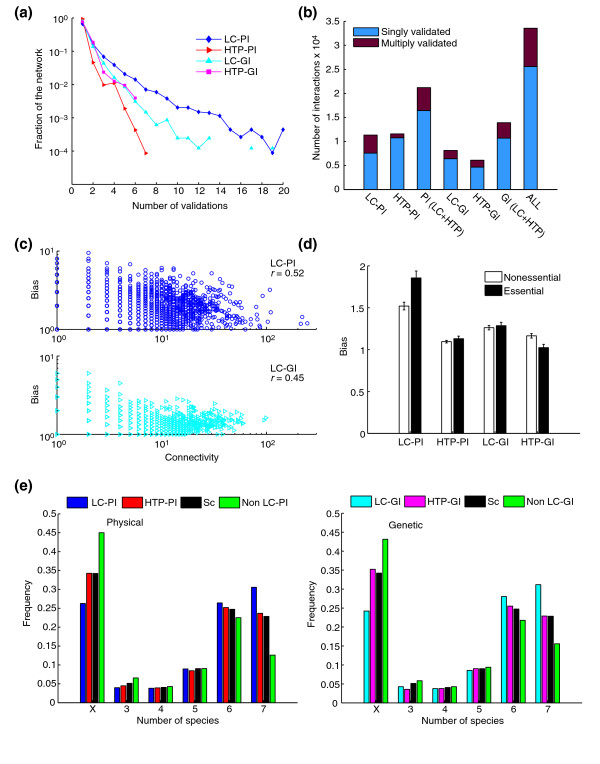
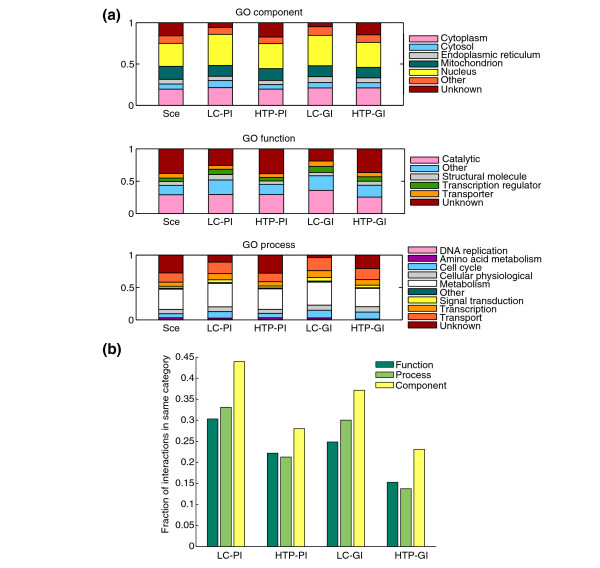
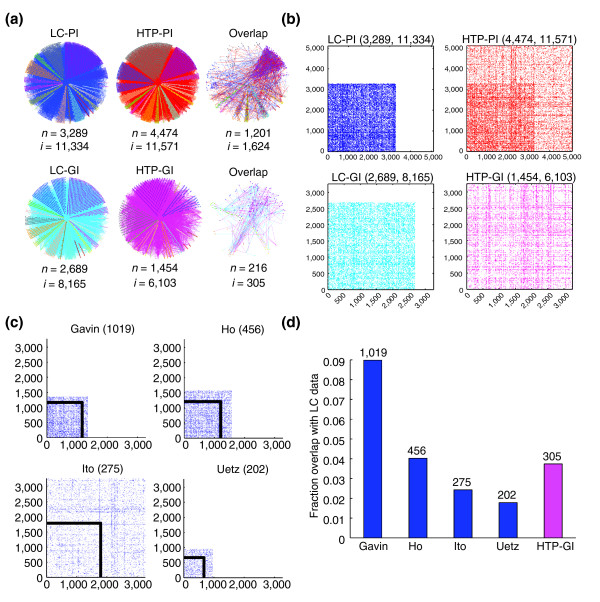
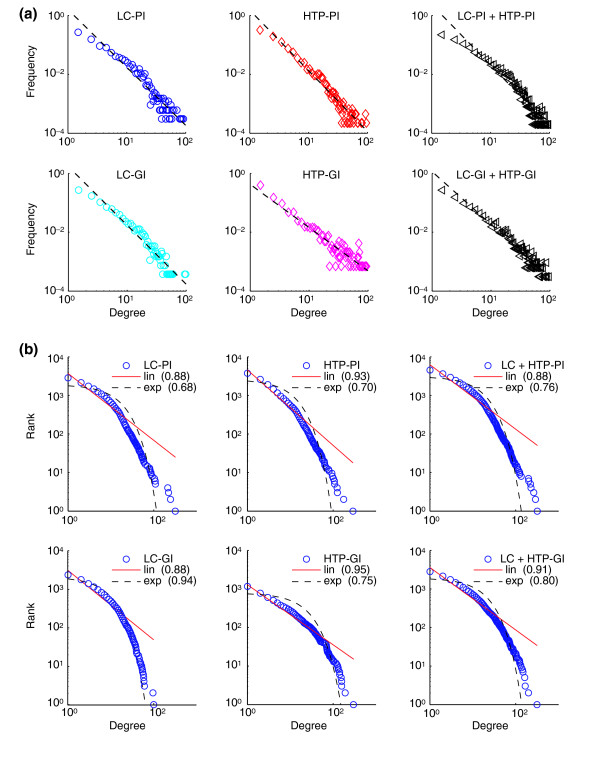
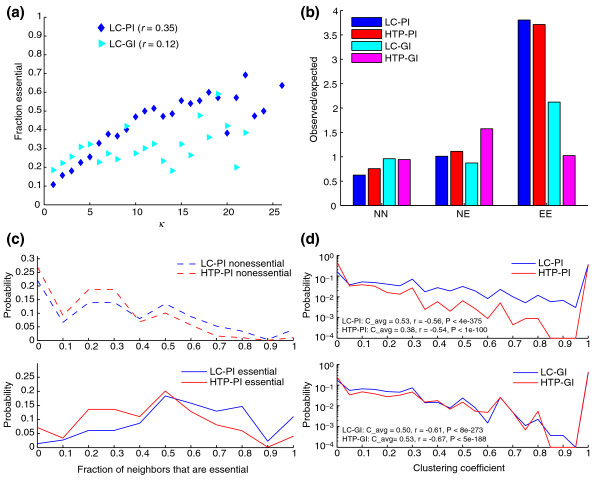
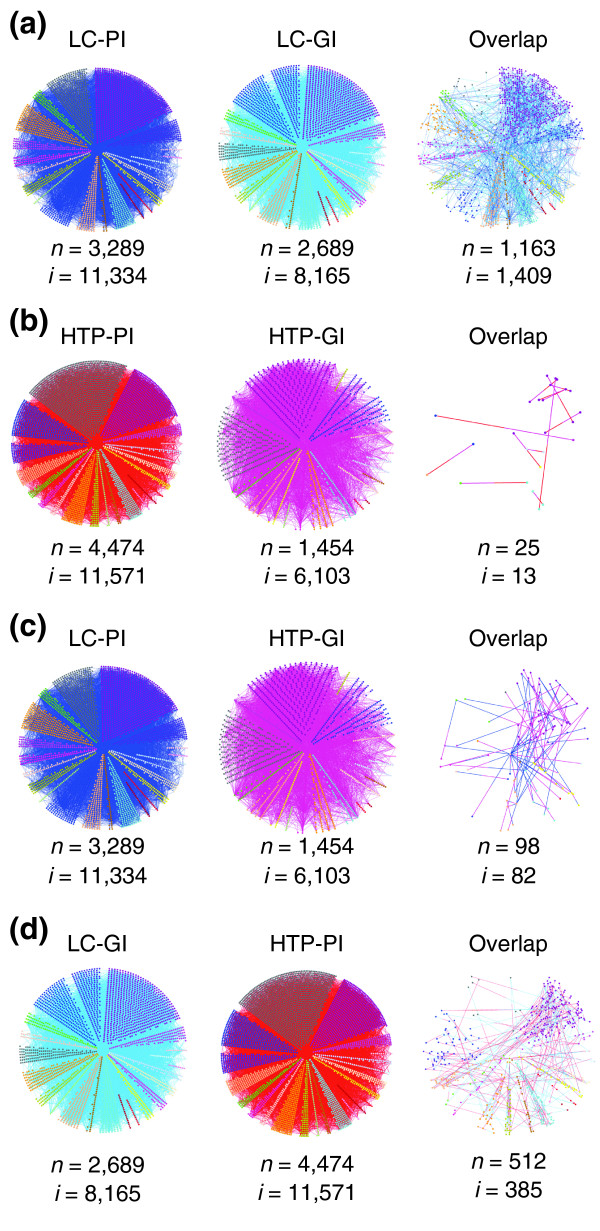
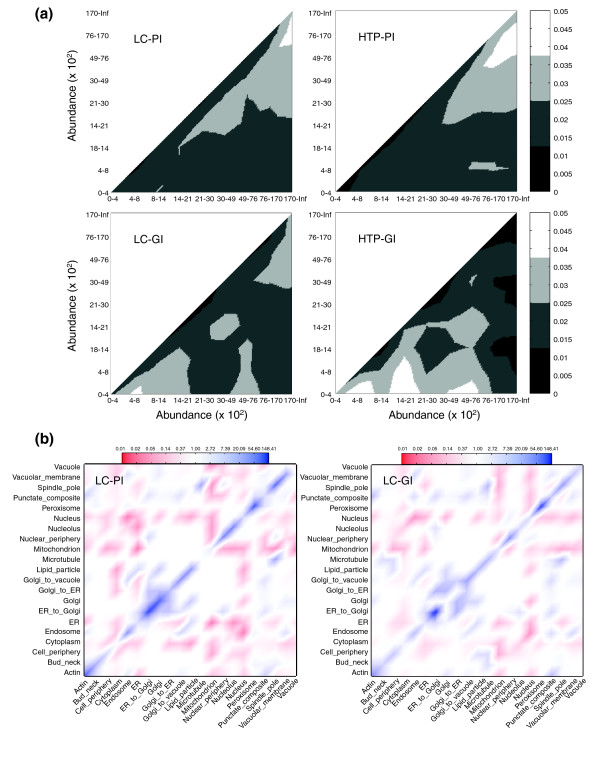
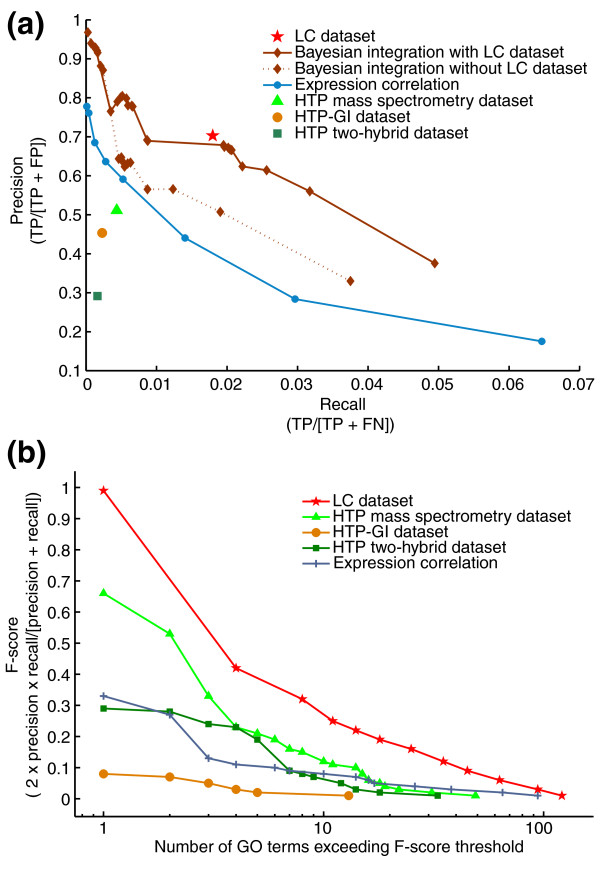
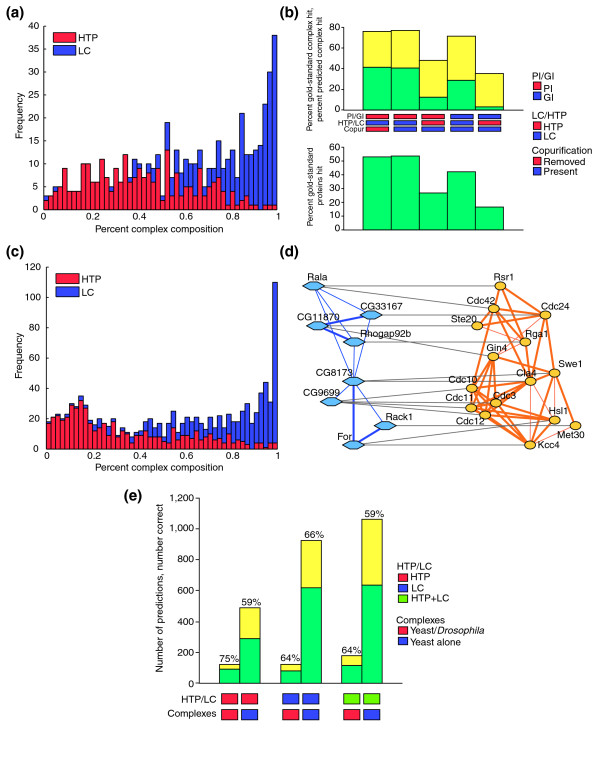
Results: We describe here a comprehensive database of genetic and protein interactions, and associated experimental evidence, for the budding yeast Saccharomyces cerevisiae, as manually curated from over 31,793 abstracts and online publications. This literature-curated (LC) dataset contains 33,311 interactions, on the order of all extant HTP datasets combined. Surprisingly, HTP protein-interaction datasets currently achieve only around 14% coverage of the interactions in the literature. The LC network nevertheless shares attributes with HTP networks, including scale-free connectivity and correlations between interactions, abundance, localization, and expression. We find that essential genes or proteins are enriched for interactions with other essential genes or proteins, suggesting that the global network may be functionally unified. This interconnectivity is supported by a substantial overlap of protein and genetic interactions in the LC dataset. We show that the LC dataset considerably improves the predictive power of network-analysis approaches. The full LC dataset is available at the BioGRID (http://www.thebiogrid.org) and SGD (http://www.yeastgenome.org/) databases.
Conclusion: Comprehensive datasets of biological interactions derived from the primary literature provide critical benchmarks for HTP methods, augment functional prediction, and reveal system-level attributes of biological networks.
Figures
Comment in
-
The interaction map of yeast: terra incognita?J Biol. 2006;5(4):10. doi: 10.1186/jbiol44. Epub 2006 Jun 8. J Biol. 2006. PMID: 16762048 Free PMC article.
Similar articles
-
The BioGRID Interaction Database: 2008 update.Nucleic Acids Res. 2008 Jan;36(Database issue):D637-40. doi: 10.1093/nar/gkm1001. Epub 2007 Nov 13. Nucleic Acids Res. 2008. PMID: 18000002 Free PMC article.
-
BioGRID: a general repository for interaction datasets.Nucleic Acids Res. 2006 Jan 1;34(Database issue):D535-9. doi: 10.1093/nar/gkj109. Nucleic Acids Res. 2006. PMID: 16381927 Free PMC article.
-
BioGRID: A Resource for Studying Biological Interactions in Yeast.Cold Spring Harb Protoc. 2016 Jan 4;2016(1):pdb.top080754. doi: 10.1101/pdb.top080754. Cold Spring Harb Protoc. 2016. PMID: 26729913 Free PMC article.
-
Functional annotations for the Saccharomyces cerevisiae genome: the knowns and the known unknowns.Trends Microbiol. 2009 Jul;17(7):286-94. doi: 10.1016/j.tim.2009.04.005. Epub 2009 Jul 2. Trends Microbiol. 2009. PMID: 19577472 Free PMC article. Review.
-
Interaction networks: lessons from large-scale studies in yeast.Proteomics. 2009 Oct;9(20):4799-811. doi: 10.1002/pmic.200900177. Proteomics. 2009. PMID: 19743423 Review.
Cited by
-
Mitotic exit and separation of mother and daughter cells.Genetics. 2012 Dec;192(4):1165-202. doi: 10.1534/genetics.112.145516. Genetics. 2012. PMID: 23212898 Free PMC article. Review.
-
Protein complexes predictions within protein interaction networks using genetic algorithms.BMC Bioinformatics. 2016 Jul 25;17 Suppl 7(Suppl 7):269. doi: 10.1186/s12859-016-1096-4. BMC Bioinformatics. 2016. PMID: 27454228 Free PMC article.
-
Crosstalk between transcription factors and microRNAs in human protein interaction network.BMC Syst Biol. 2012 Mar 13;6:18. doi: 10.1186/1752-0509-6-18. BMC Syst Biol. 2012. PMID: 22413876 Free PMC article.
-
Counting motifs in the human interactome.Nat Commun. 2013;4:2241. doi: 10.1038/ncomms3241. Nat Commun. 2013. PMID: 23917172 Free PMC article.
-
Comparative interaction networks: bridging genotype to phenotype.Adv Exp Med Biol. 2012;751:139-56. doi: 10.1007/978-1-4614-3567-9_7. Adv Exp Med Biol. 2012. PMID: 22821457 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
