

IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice
- PMID: 16717119
- PMCID: PMC2118311
- DOI: 10.1084/jem.20060436
IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice
Abstract
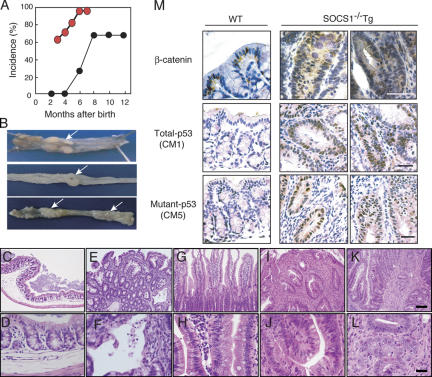
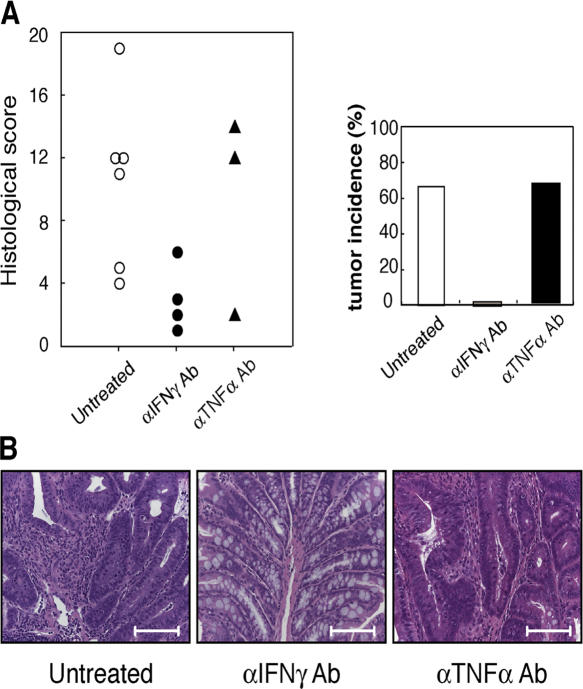
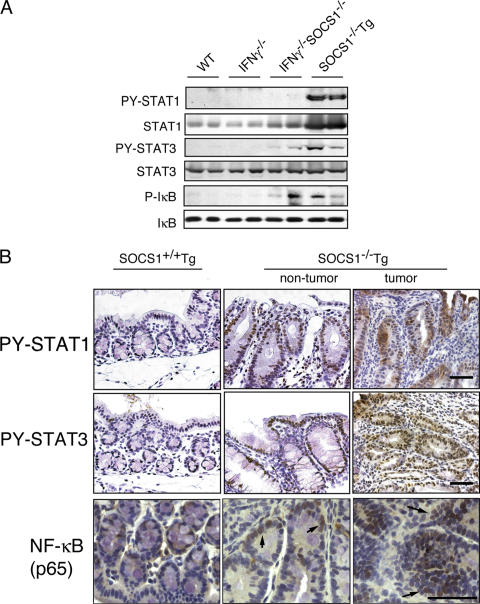
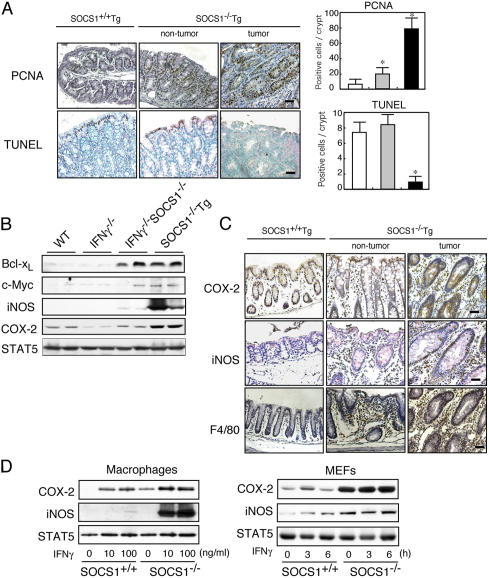
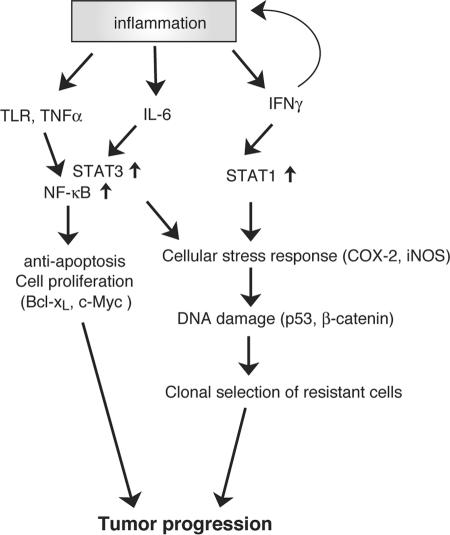
Approximately 20% of human cancers are estimated to develop from chronic inflammation. Recently, the NF-kappaB pathway was shown to play an essential role in promoting inflammation-associated cancer, but the role of the JAK/STAT pathway, another important signaling pathway of proinflammatory cytokines, remains to be investigated. Suppressor of cytokine signaling-1 (SOCS1) acts as an important physiological regulator of cytokine responses, and silencing of the SOCS1 gene by DNA methylation has been found in several human cancers. Here, we demonstrated that SOCS1-deficient mice (SOCS1-/- Tg mice), in which SOCS1 expression was restored in T and B cells on a SOCS1-/- background, spontaneously developed colorectal carcinomas carrying nuclear beta-catenin accumulation and p53 mutations at 6 months of age. However, interferon (IFN)gamma-/- SOCS1-/- mice and SOCS1-/- Tg mice treated with anti-IFNgamma antibody did not develop such tumors. STAT3 and NF-kappaB activation was evident in SOCS1-/- Tg mice, but these were not sufficient for tumor development because these are also activated in IFNgamma-/- SOCS1-/- mice. However, colons of SOCS1-/- Tg mice, but not IFNgamma-/- SOCS1-/- mice, showed hyperactivation of STAT1, which resulted in the induction of carcinogenesis-related enzymes, cyclooxygenase-2 and inducible nitric oxide synthase. These data strongly suggest that SOCS1 is a unique antioncogene which prevents chronic inflammation-mediated carcinogenesis by regulation of the IFNgamma/STAT1 pathways.
Figures
Similar articles
-
Suppressor of cytokine signaling-1 regulates inflammatory bowel disease in which both IFNgamma and IL-4 are involved.Gastroenterology. 2006 Feb;130(2):373-88. doi: 10.1053/j.gastro.2005.10.051. Gastroenterology. 2006. PMID: 16472593
-
Loss of suppressor of cytokine signaling 1 in helper T cells leads to defective Th17 differentiation by enhancing antagonistic effects of IFN-gamma on STAT3 and Smads.J Immunol. 2008 Mar 15;180(6):3746-56. doi: 10.4049/jimmunol.180.6.3746. J Immunol. 2008. PMID: 18322180
-
Acute alcohol intake induces SOCS1 and SOCS3 and inhibits cytokine-induced STAT1 and STAT3 signaling in human monocytes.Alcohol Clin Exp Res. 2008 Sep;32(9):1565-73. doi: 10.1111/j.1530-0277.2008.00726.x. Epub 2008 Jul 9. Alcohol Clin Exp Res. 2008. PMID: 18616672 Free PMC article.
-
SOCS1: a potent and multifaceted regulator of cytokines and cell-mediated inflammation.Tissue Antigens. 2006 Jan;67(1):1-9. doi: 10.1111/j.1399-0039.2005.00532.x. Tissue Antigens. 2006. PMID: 16451196 Review.
-
Role of SOCS1 in tumor progression and therapeutic application.Int J Cancer. 2012 May 1;130(9):1971-80. doi: 10.1002/ijc.27318. Epub 2012 Jan 11. Int J Cancer. 2012. PMID: 22025331 Review.
Cited by
-
Protein-bound polysaccharide-K reduces colitic tumors and improves survival of inflammatory bowel disease in vivo.Oncol Lett. 2011 Sep 1;2(5):791-796. doi: 10.3892/ol.2011.336. Epub 2011 Jul 4. Oncol Lett. 2011. PMID: 22866128 Free PMC article.
-
[The Role of SOCS in the Development of Tumors].Zhongguo Fei Ai Za Zhi. 2016 Sep 20;19(9):620-5. doi: 10.3779/j.issn.1009-3419.2016.09.11. Zhongguo Fei Ai Za Zhi. 2016. PMID: 27666555 Free PMC article. Review. Chinese.
-
Identification and characterization of suppressor of cytokine signaling 1 (SOCS-1) homologues in teleost fish.Immunogenetics. 2007 Aug;59(8):673-86. doi: 10.1007/s00251-007-0232-8. Epub 2007 Jun 14. Immunogenetics. 2007. PMID: 17569039
-
Knockdown of miR-92a suppresses the stemness of colorectal cancer cells via mediating SOCS3.Bioengineered. 2022 Mar;13(3):5613-5624. doi: 10.1080/21655979.2021.2022267. Bioengineered. 2022. PMID: 35184640 Free PMC article.
-
Immunotherapy for Colorectal Cancer: Mechanisms and Predictive Biomarkers.Cancers (Basel). 2022 Feb 17;14(4):1028. doi: 10.3390/cancers14041028. Cancers (Basel). 2022. PMID: 35205776 Free PMC article. Review.
References
-
- Itzkowitz, S.H., and X. Yio. 2004. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 287:G7–G17. - PubMed
-
- Oshima, M., and M.M. Taketo. 2002. COX selectivity and animal models for colon cancer. Curr. Pharm. Des. 8:1021–1034. - PubMed
-
- Greten, F.R., L. Eckmann, T.F. Greten, J.M. Park, Z.W. Li, L.J. Egan, M.F. Kagnoff, and M. Karin. 2004. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 118:285–296. - PubMed
-
- Pikarsky, E., R.M. Porat, I. Stein, R. Abramovitch, S. Amit, S. Kasem, E. Gutkovich-Pyest, S. Urieli-Shoval, E. Galun, and Y. Ben-Neriah. 2004. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 431:461–466. - PubMed
-
- Fiocchi, C. 1998. Inflammatory bowel disease: etiology and pathogenesis. Gastroenterology. 115:182–205. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
