

Multipass membrane protein structure prediction using Rosetta
- PMID: 16372357
- PMCID: PMC1479309
- DOI: 10.1002/prot.20817
Multipass membrane protein structure prediction using Rosetta
Abstract
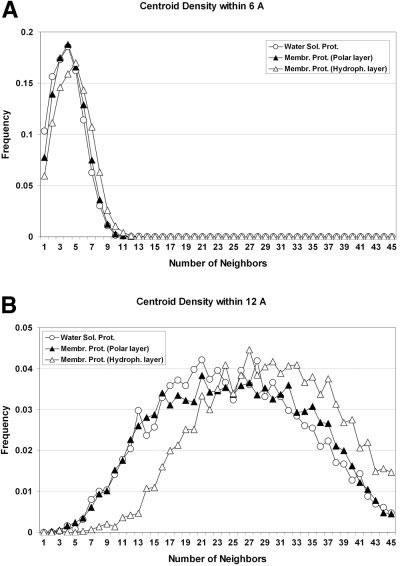
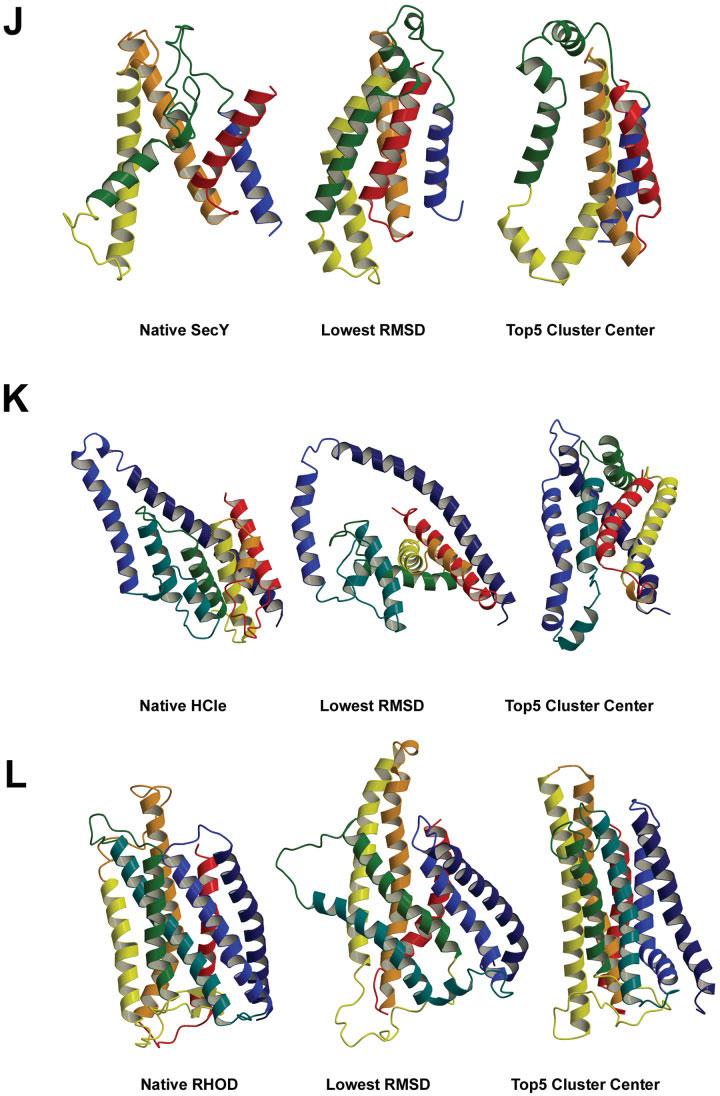
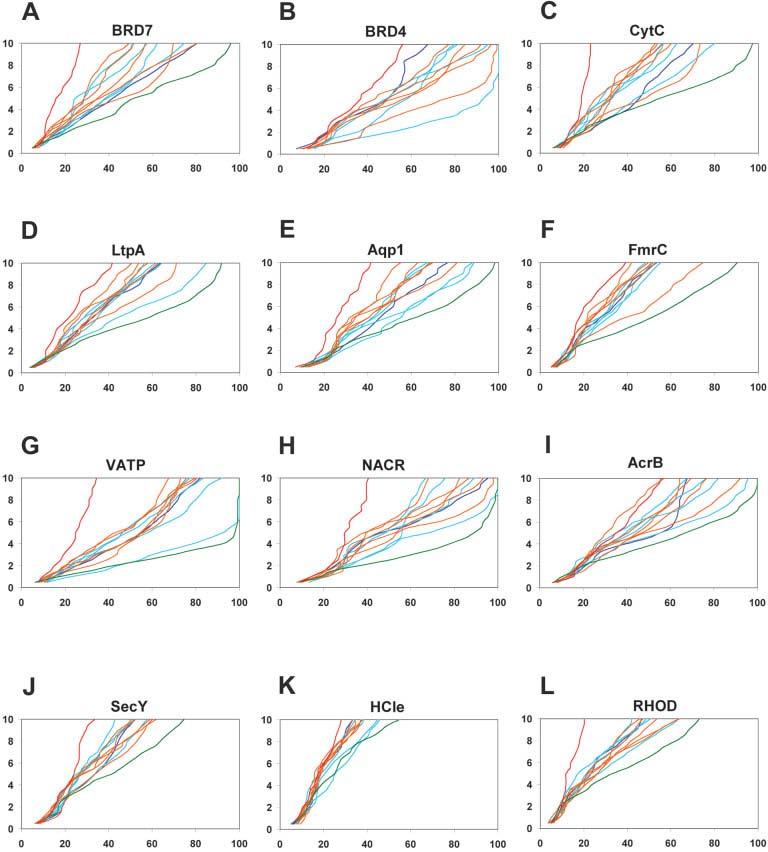
We describe the adaptation of the Rosetta de novo structure prediction method for prediction of helical transmembrane protein structures. The membrane environment is modeled by embedding the protein chain into a model membrane represented by parallel planes defining hydrophobic, interface, and polar membrane layers for each energy evaluation. The optimal embedding is determined by maximizing the exposure of surface hydrophobic residues within the membrane and minimizing hydrophobic exposure outside of the membrane. Protein conformations are built up using the Rosetta fragment assembly method and evaluated using a new membrane-specific version of the Rosetta low-resolution energy function in which residue-residue and residue-environment interactions are functions of the membrane layer in addition to amino acid identity, distance, and density. We find that lower energy and more native-like structures are achieved by sequential addition of helices to a growing chain, which may mimic some aspects of helical protein biogenesis after translocation, rather than folding the whole chain simultaneously as in the Rosetta soluble protein prediction method. In tests on 12 membrane proteins for which the structure is known, between 51 and 145 residues were predicted with root-mean-square deviation <4 A from the native structure.
2005 Wiley-Liss, Inc.
Figures





Similar articles
-
Improving fragment quality for de novo structure prediction.Proteins. 2014 Sep;82(9):2240-52. doi: 10.1002/prot.24587. Epub 2014 May 2. Proteins. 2014. PMID: 24753351
-
Comparison of helix interactions in membrane and soluble alpha-bundle proteins.Biophys J. 2002 May;82(5):2720-36. doi: 10.1016/S0006-3495(02)75613-0. Biophys J. 2002. PMID: 11964258 Free PMC article.
-
Systematic evaluation of CS-Rosetta for membrane protein structure prediction with sparse NOE restraints.Proteins. 2017 May;85(5):812-826. doi: 10.1002/prot.25224. Epub 2017 Feb 28. Proteins. 2017. PMID: 27936510
-
Practically useful: what the Rosetta protein modeling suite can do for you.Biochemistry. 2010 Apr 13;49(14):2987-98. doi: 10.1021/bi902153g. Biochemistry. 2010. PMID: 20235548 Free PMC article. Review.
-
Membrane enzymes: transformers at the interface.Nat Chem Biol. 2015 Feb;11(2):102-3. doi: 10.1038/nchembio.1738. Epub 2015 Jan 12. Nat Chem Biol. 2015. PMID: 25580855 Review. No abstract available.
Cited by
-
Replica exchange Monte-Carlo simulations of helix bundle membrane proteins: rotational parameters of helices.J Comput Aided Mol Des. 2012 Mar;26(3):363-74. doi: 10.1007/s10822-012-9562-1. Epub 2012 Mar 31. J Comput Aided Mol Des. 2012. PMID: 22466784
-
Toward high-resolution computational design of the structure and function of helical membrane proteins.Nat Struct Mol Biol. 2016 Jun 7;23(6):475-80. doi: 10.1038/nsmb.3231. Nat Struct Mol Biol. 2016. PMID: 27273630 Free PMC article.
-
Structure determination of a membrane protein in proteoliposomes.J Am Chem Soc. 2012 Feb 1;134(4):2047-56. doi: 10.1021/ja209464f. Epub 2012 Jan 23. J Am Chem Soc. 2012. PMID: 22217388 Free PMC article.
-
Recent Advances in the Application of Solution NMR Spectroscopy to Multi-Span Integral Membrane Proteins.Prog Nucl Magn Reson Spectrosc. 2009 Nov 1;55(4):335-360. doi: 10.1016/j.pnmrs.2009.07.002. Prog Nucl Magn Reson Spectrosc. 2009. PMID: 20161395 Free PMC article. No abstract available.
-
Ion channel voltage sensors: structure, function, and pathophysiology.Neuron. 2010 Sep 23;67(6):915-28. doi: 10.1016/j.neuron.2010.08.021. Neuron. 2010. PMID: 20869590 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
