

Fitting background-selection predictions to levels of nucleotide variation and divergence along the human autosomes
- PMID: 16140989
- PMCID: PMC1199535
- DOI: 10.1101/gr.3413205
Fitting background-selection predictions to levels of nucleotide variation and divergence along the human autosomes
Abstract
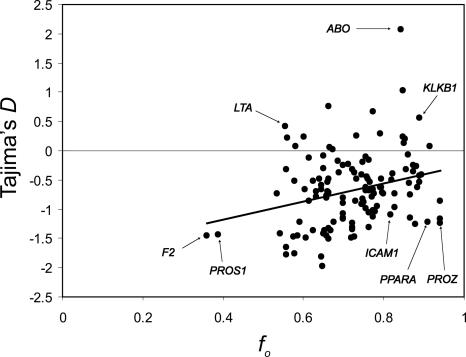
The roles of positive directional selection (selective sweeps) and negative selection (background selection) in shaping the genome-wide distribution of genetic variation in humans remain largely unknown. Here, we optimize the parameter values of a model of the removal of deleterious mutations (background selection) to observed levels of human polymorphism, controlling for mutation rate heterogeneity by using interspecific divergence. A point of "best fit" was found between background-selection predictions and estimates of human effective population sizes, with reasonable parameter estimates whose uncertainty was assessed by bootstrapping. The results suggest that the purging of deleterious alleles has had some influence on shaping levels of human variation, although the effects may be subtle over the majority of the human genome. A significant relationship was found between background-selection predictions and measures of skew in the allele frequency distribution. The genome-wide action of selection (positive and/or negative) is required to explain this observation.
Figures


Similar articles
-
Searching for evidence of positive selection in the human genome using patterns of microsatellite variability.Mol Biol Evol. 2002 Jul;19(7):1143-53. doi: 10.1093/oxfordjournals.molbev.a004172. Mol Biol Evol. 2002. PMID: 12082133
-
Broad-scale variation in human genetic diversity levels is predicted by purifying selection on coding and non-coding elements.Elife. 2023 Jun 23;12:e76065. doi: 10.7554/eLife.76065. Elife. 2023. PMID: 36196994 Free PMC article.
-
Natural selection affects multiple aspects of genetic variation at putatively neutral sites across the human genome.PLoS Genet. 2011 Oct;7(10):e1002326. doi: 10.1371/journal.pgen.1002326. Epub 2011 Oct 13. PLoS Genet. 2011. PMID: 22022285 Free PMC article.
-
Positive natural selection in the human lineage.Science. 2006 Jun 16;312(5780):1614-20. doi: 10.1126/science.1124309. Science. 2006. PMID: 16778047 Review.
-
Signatures of positive selection: from selective sweeps at individual loci to subtle allele frequency changes in polygenic adaptation.Mol Ecol. 2016 Jan;25(1):79-88. doi: 10.1111/mec.13288. Epub 2015 Jul 27. Mol Ecol. 2016. PMID: 26108992 Review.
Cited by
-
Variable Autosomal and X Divergence Near and Far from Genes Affects Estimates of Male Mutation Bias in Great Apes.Genome Biol Evol. 2016 Dec 31;8(11):3393-3405. doi: 10.1093/gbe/evw232. Genome Biol Evol. 2016. PMID: 27702816 Free PMC article.
-
The effects of deleterious mutations on evolution at linked sites.Genetics. 2012 Jan;190(1):5-22. doi: 10.1534/genetics.111.134288. Genetics. 2012. PMID: 22219506 Free PMC article. Review.
-
Fifteen years of genomewide scans for selection: trends, lessons and unaddressed genetic sources of complication.Mol Ecol. 2016 Jan;25(1):5-23. doi: 10.1111/mec.13339. Epub 2015 Sep 16. Mol Ecol. 2016. PMID: 26224644 Free PMC article. Review.
-
Patterns of variation in DNA segments upstream of transcription start sites.Hum Mutat. 2007 May;28(5):441-50. doi: 10.1002/humu.20463. Hum Mutat. 2007. PMID: 17274005 Free PMC article.
-
Determining the Effect of Natural Selection on Linked Neutral Divergence across Species.PLoS Genet. 2016 Aug 10;12(8):e1006199. doi: 10.1371/journal.pgen.1006199. eCollection 2016 Aug. PLoS Genet. 2016. PMID: 27508305 Free PMC article.
References
-
- Andolfatto, P. 2001. Adaptive hitchhiking effects on genome variability. Curr. Opin. Genet. Dev. 11: 635-641. - PubMed
-
- Antezana, M.A. and Hudson, R.R. 1997. Before crossing over: The advantages of eukaryotic sex in genomes lacking chiasmatic recombination. Genet. Res. 70: 7-25. - PubMed
-
- Aquadro, C.F., Bauer DuMont, V., and Reed, F.A. 2001. Genome-wide variation in the human and fruitfly: A comparison. Curr. Opin. Genet. Dev. 11: 627-634. - PubMed
WEB SITE REFERENCES
-
- http://genome.ucsc.edu/; UCSC Human Genome Browser. - PubMed
-
- http://pga.gs.washington.edu; SeattleSNPs, NHLBI Program for Genomic Applications, UW-FHCRC, Seattle, WA, March 26, 2005.
-
- http://www.ncbi.nlm.nih.gov/omim/; Online Mendelian Inheritance in Man (OMIM).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources