

Review
doi: 10.1101/sqb.2004.69.447.
The new field of epigenomics: implications for cancer and other common disease research
Affiliations
- PMID: 16117680
- PMCID: PMC5434869
- DOI: 10.1101/sqb.2004.69.447
Item in Clipboard
Review
The new field of epigenomics: implications for cancer and other common disease research
Cold Spring Harb Symp Quant Biol.
2004.
No abstract available
Figures
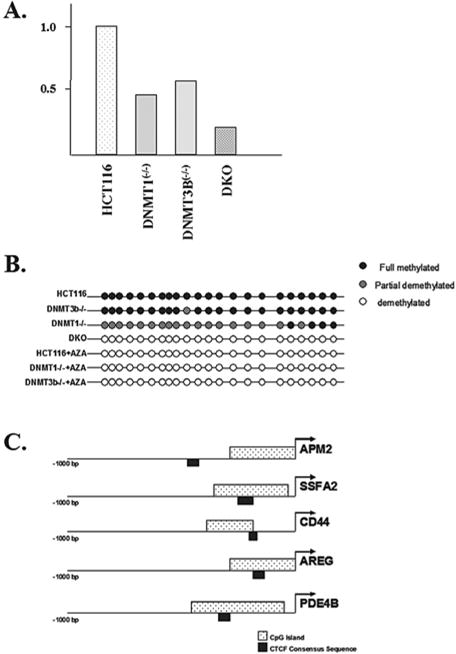
Expression of the APM2 gene is decreased in somatic cell methyltransferase knockout cell lines. (A) The total RNA was isolated from control HCT116, as well as dmnt1(−/−),dnmt3B(−/−), and double knockout (DKO) cells and APM2 RNA levels were determined via real-time RT-PCR (reverse transcriptase polymerase chain reaction). Results represent the average of three independent reactions. (B) The APM2 promoter CpG island is hypomethylated in somatic cell methyltransferase knockout cell lines. The methylation status of the CpG island in the APM2 upstream promoter region was determined in HCT116, DNMT1(−/−), DNMT3B(−/−), and DKO cells. (C) The APM2, SSFA2, CD44, AREG, and PDE4B promoters were examined for the presence of CpG islands and CTCF binding sites using software available at Entrez Genome (http://ncbi.nhn.nih.gov ).
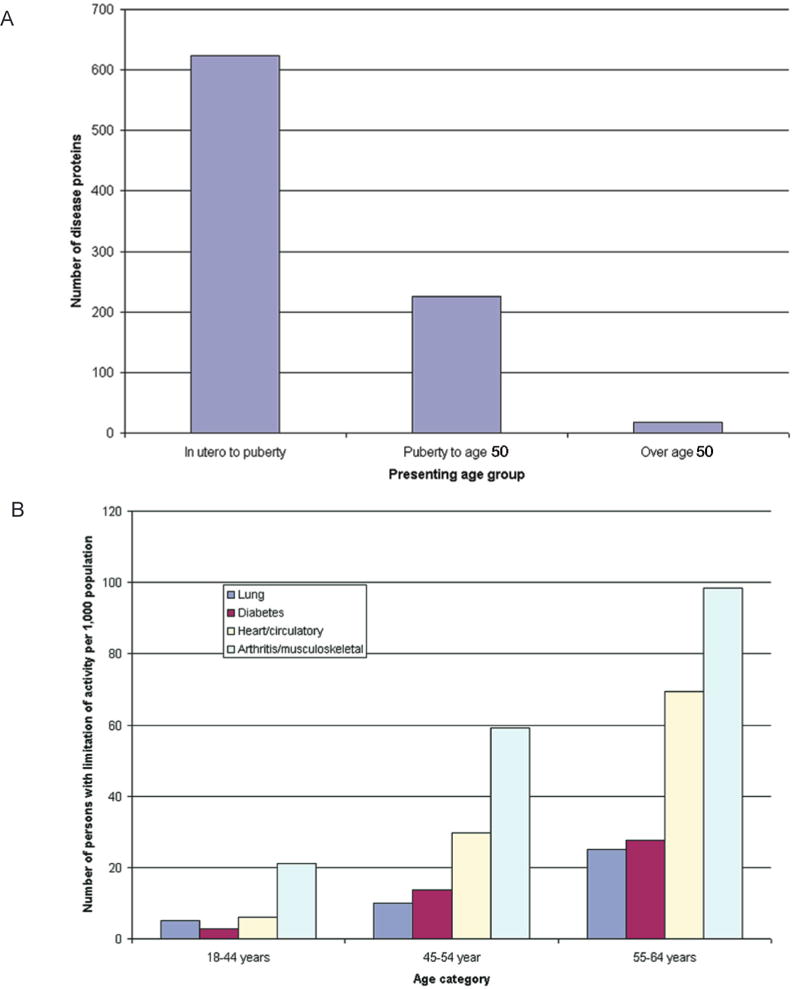
Age of presentation of conventional (A) Mendelian disorders and (B) common complex traits causing limitation of activity among working-age adults, 1999–2001. Data for panel A are from Jimenez-Sanchez et al. (2001), and data for panel B are from the U.S. National Center for Health Statistics (Statistics 2003). (Reprinted, with permission, from Bjornsson et al. 2004 [©Elsevier].)
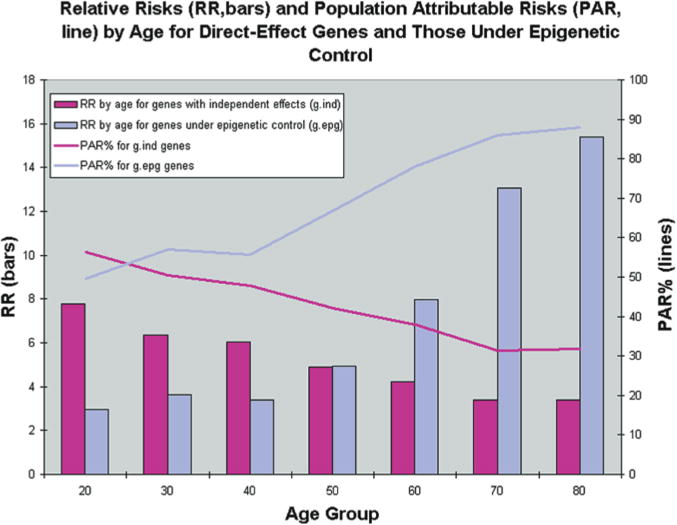
Results from simulations of 40 populations. Simulations were used to create 40 populations containing affected and unaffected individuals for a genetic epidemiologic analysis. Age, environmental status, and genotypes for three different genes were first simulated at random according to specified frequencies. Disease status was then simulated according to CDGE. From these simulated populations, relative risks for gind and gdep genes were estimated in cross-sectional analysis by each age decade, and averaged over 40 populations (bars). Because this risk reflects only the magnitude of a genetic effect, and not the importance of that genotype with respect to all cases in the population, we also estimated population attributable risk percentage (PAR%) at each decade. This reflects the proportion of cases in the population that can be explained by the particular genetic effect. (Reprinted, with permission, from Bjornsson et al. 2004 [©Elsevier].)
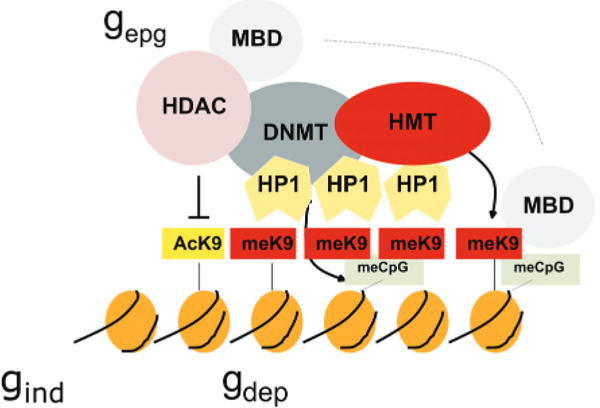
Cooperative and self-reinforcing organization of the chromatin- and DNA-modifying machinery responsible for gene silencing in normal and malignant cells. Histone (H3) modifications include lysine (K) acetylation (Ac) and lysine methylation (Me). Lysines at other positions are also modified. The HP1 protein recognizes MeK9 and, as this protein also binds the histone methyltransferase (HMT), heterochromatin can spread. Histone deacetylases (HDAC) deacetylate lysine residues as a prerequisite for their subsequent methylation. DNA methyltransferases (DNMT) participate in multiprotein complexes that contain HDACs and HMTs, and methyl-C binding proteins (MBD) can be loaded onto methylated DNA through their interactions with both HDACs and HMTs. Much of the evidence comes from studies of constitutive heterochromatin, but recent studies indicate similar interactions of genes silenced de novo in cancer cells. (Reprinted, with permission, from Feinberg and Tycko 2004 [©Nature Publishing Group; http://www.nature.com ].)
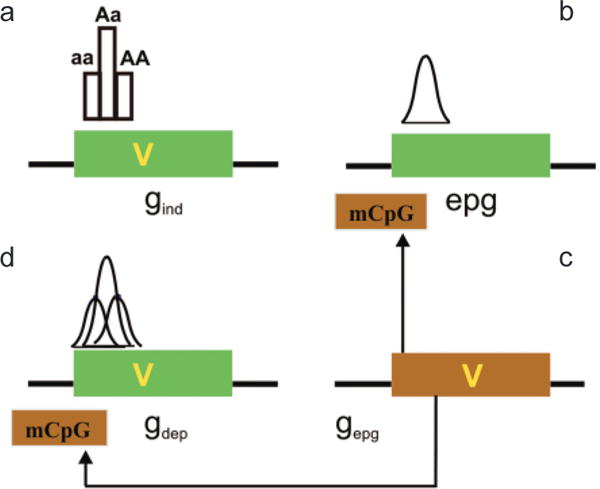
Interaction of genetic and epigenetic variation at a genomic level. (a) A gene (green box) can contain a sequence variant (V) that contributes to disease. Gene variants that are not influenced by epigenetic modification in their disease contribution (although epigenetics can contribute to normal function) are epigenetic independent (gind). The distribution of phenotype of a single locus will not be Gaussian in the absence of other factors, e.g., environmental. (b) Epigenetic variation can contribute to disease phenotype directly, independent of a genetic variation in the target gene (epg). Epigenetic variation itself is quantitative and thus can impart the quantitative nature to a trait, even at a single locus. (c) If the penetrance of a gene sequence variant is affected by epigenetic modification (mCpG), the gene is “epigenetic dependent” (gdep). In this case, the genetic and epigenetic variation together could contribute to a Gaussian distribution, even at a single locus. Note that the epigenetic modification is drawn on the gene but it could be at some distance upstream or downstream from that gene. The epigenetic modification need not be methylation, which is drawn here for convenience. (d) A genetic variant that can influence this epigenetic modification (e.g., encoding a chromatin-modifying protein) is referred to as gepg, and its influence is denoted by arrows. (Reprinted, with permission, from Bjornsson et al. 2004 [©Elsevier].)
Similar articles
-
The complexity of epigenetic diseases.J Pathol. 2016 Jan;238(2):333-44. doi: 10.1002/path.4647. Epub 2015 Nov 17. J Pathol. 2016. PMID: 26419725 Free PMC article. Review.
-
In brief: genomic imprinting and imprinting diseases.J Pathol. 2014 Apr;232(5):485-7. doi: 10.1002/path.4326. Epub 2014 Jan 29. J Pathol. 2014. PMID: 24395592 Review.
-
Mechanisms of disease: epigenesis.Semin Pediatr Neurol. 2007 Mar;14(1):7-14. doi: 10.1016/j.spen.2006.11.004. Semin Pediatr Neurol. 2007. PMID: 17331879 Review.
-
The history of cancer epigenetics.Nat Rev Cancer. 2004 Feb;4(2):143-53. doi: 10.1038/nrc1279. Nat Rev Cancer. 2004. PMID: 14732866 Review. No abstract available.
-
Gene silencing.N Engl J Med. 2004 Feb 26;350(9):947-8; author reply 947-8. doi: 10.1056/NEJM200402263500920. N Engl J Med. 2004. PMID: 14985497 No abstract available.
Cited by
-
Epigenetics: deciphering how environmental factors may modify autoimmune type 1 diabetes.Mamm Genome. 2009 Sep-Oct;20(9-10):624-32. doi: 10.1007/s00335-009-9213-6. Epub 2009 Aug 22. Mamm Genome. 2009. PMID: 19697079 Review.
-
Altered DNA methylation patterns of the H19 differentially methylated region and the DAZL gene promoter are associated with defective human sperm.PLoS One. 2013 Aug 28;8(8):e71215. doi: 10.1371/journal.pone.0071215. eCollection 2013. PLoS One. 2013. PMID: 24015185 Free PMC article.
-
Infant siblings and the investigation of autism risk factors.J Neurodev Disord. 2012 Apr 18;4(1):7. doi: 10.1186/1866-1955-4-7. J Neurodev Disord. 2012. PMID: 22958474 Free PMC article.
-
Transcriptomic and genomic analysis of human hepatocellular carcinomas and hepatoblastomas.Hepatology. 2006 Oct;44(4):1012-24. doi: 10.1002/hep.21328. Hepatology. 2006. PMID: 17006932 Free PMC article.
-
Characterization of the DNA methylome and its interindividual variation in human peripheral blood monocytes.Epigenomics. 2013 Jun;5(3):255-69. doi: 10.2217/epi.13.18. Epigenomics. 2013. PMID: 23750642 Free PMC article.
References
-
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185. - PubMed
-
- Bachman KE, Park BH, Rhee I, Rajagopalan H, Herman JG, Baylin SB, Kinzler KW, Vogelstein B. Histone modifications and silencing prior to DNA methylation of a tumor suppressor gene. Cancer Cell. 2003;3:89. - PubMed
-
- Bakin AV, Curran T. Role of DNA 5-methylcytosine transferase in cell transformation by fos. Science. 1999;283:387. - PubMed
-
- Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9:2395. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources