

B cells from hyper-IgM patients carrying UNG mutations lack ability to remove uracil from ssDNA and have elevated genomic uracil
- PMID: 15967827
- PMCID: PMC2212036
- DOI: 10.1084/jem.20050042
B cells from hyper-IgM patients carrying UNG mutations lack ability to remove uracil from ssDNA and have elevated genomic uracil
Abstract
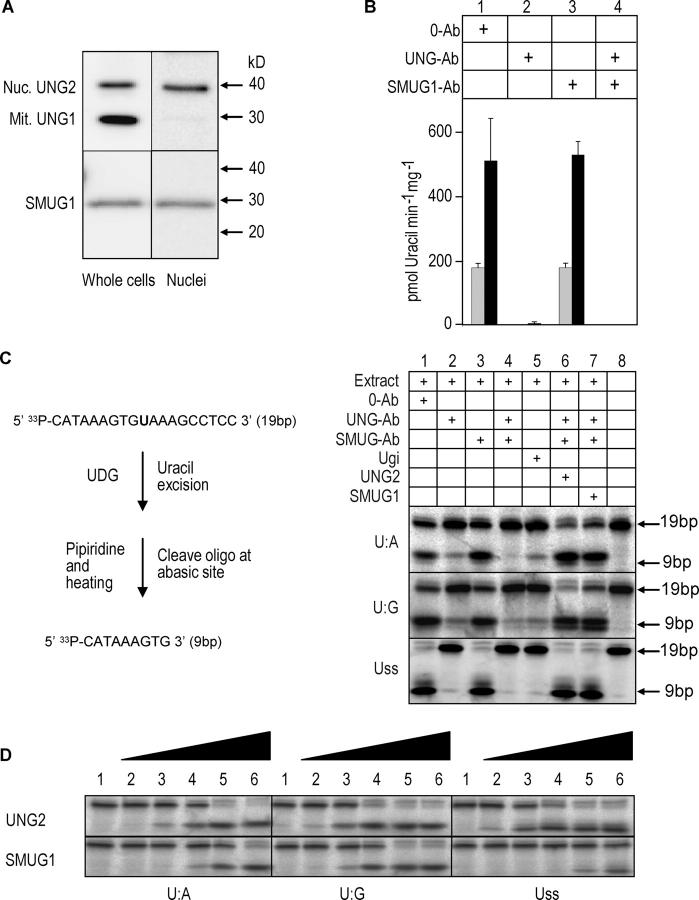
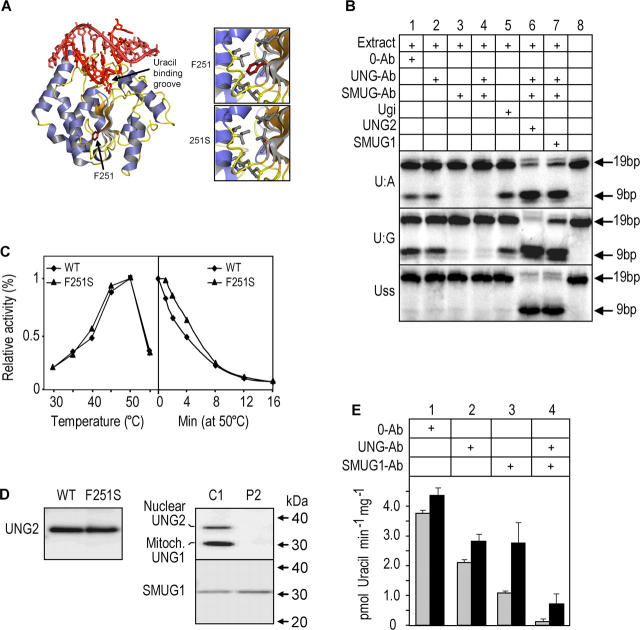
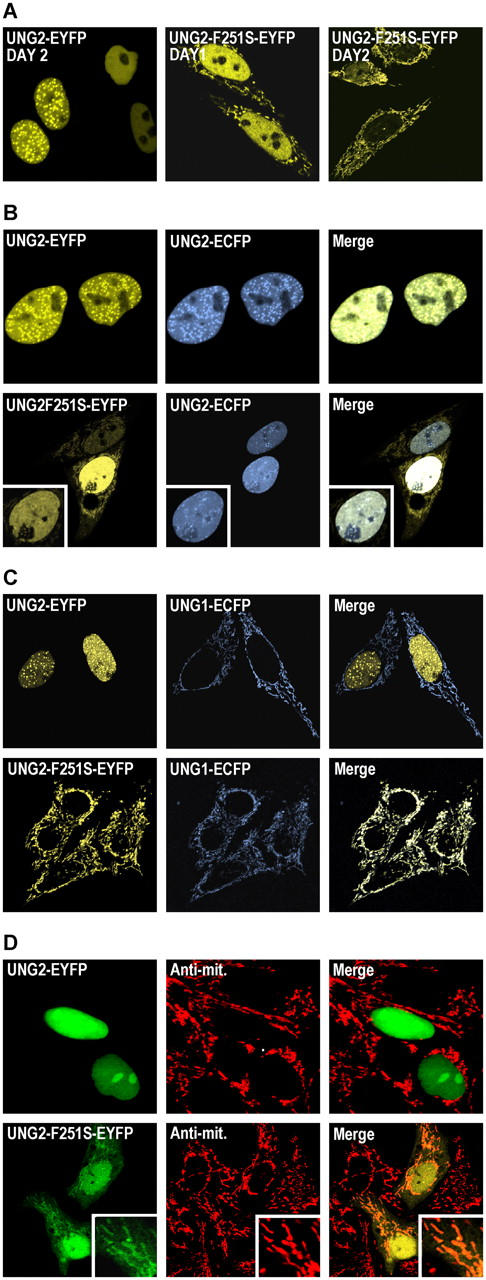
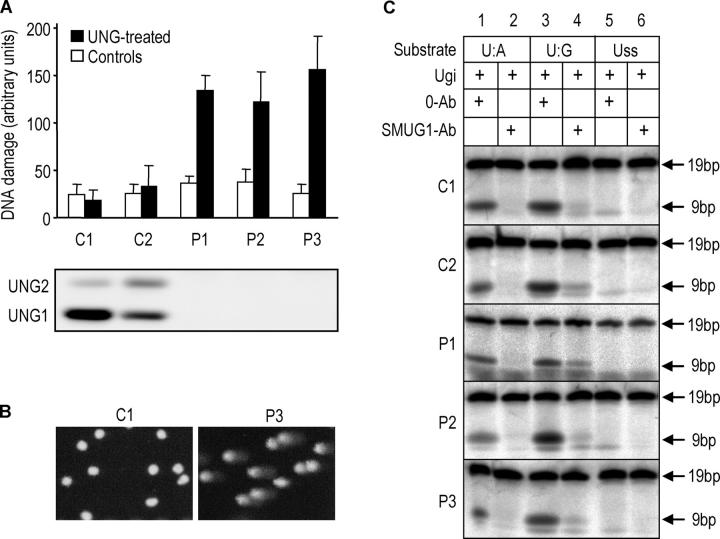
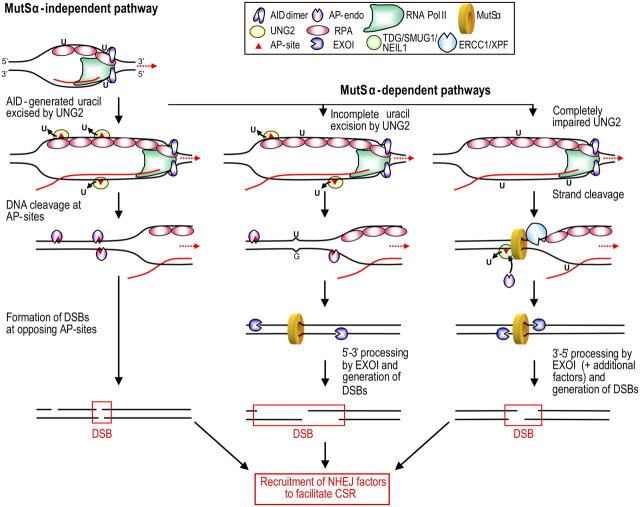
The generation of high-affinity antibodies requires somatic hypermutation (SHM) and class switch recombination (CSR) at the immunoglobulin (Ig) locus. Both processes are triggered by activation-induced cytidine deaminase (AID) and require UNG-encoded uracil-DNA glycosylase. AID has been suggested to function as an mRNA editing deaminase or as a single-strand DNA deaminase. In the latter model, SHM may result from replicative incorporation of dAMP opposite U or from error-prone repair of U, whereas CSR may be triggered by strand breaks at abasic sites. Here, we demonstrate that extracts of UNG-proficient human B cell lines efficiently remove U from single-stranded DNA. In B cell lines from hyper-IgM patients carrying UNG mutations, the single-strand-specific uracil-DNA glycosylase, SMUG1, cannot complement this function. Moreover, the UNG mutations lead to increased accumulation of genomic uracil. One mutation results in an F251S substitution in the UNG catalytic domain. Although this UNG form was fully active and stable when expressed in Escherichia coli, it was mistargeted to mitochondria and degraded in mammalian cells. Our results may explain why SMUG1 cannot compensate the UNG2 deficiency in human B cells, and are fully consistent with the DNA deamination model that requires active nuclear UNG2. Based on our findings and recent information in the literature, we present an integrated model for the initiating steps in CSR.
Figures
Similar articles
-
Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination.Nat Immunol. 2003 Oct;4(10):1023-8. doi: 10.1038/ni974. Epub 2003 Sep 7. Nat Immunol. 2003. PMID: 12958596
-
Uracil in DNA--general mutagen, but normal intermediate in acquired immunity.DNA Repair (Amst). 2007 Apr 1;6(4):505-16. doi: 10.1016/j.dnarep.2006.10.014. Epub 2006 Nov 20. DNA Repair (Amst). 2007. PMID: 17116429 Review.
-
Genomic uracil and human disease.Exp Cell Res. 2006 Aug 15;312(14):2666-72. doi: 10.1016/j.yexcr.2006.06.015. Epub 2006 Jun 21. Exp Cell Res. 2006. PMID: 16860315 Review.
-
Single-strand DNA breaks in Ig class switch recombination that depend on UNG but not AID.Int Immunol. 2008 Nov;20(11):1381-93. doi: 10.1093/intimm/dxn097. Epub 2008 Sep 15. Int Immunol. 2008. PMID: 18794203
-
Uracil DNA glycosylase activity is dispensable for immunoglobulin class switch.Science. 2004 Aug 20;305(5687):1160-3. doi: 10.1126/science.1098444. Science. 2004. PMID: 15326357
Cited by
-
APOBEC3G enhances lymphoma cell radioresistance by promoting cytidine deaminase-dependent DNA repair.Blood. 2012 Jul 12;120(2):366-75. doi: 10.1182/blood-2012-01-402123. Epub 2012 May 29. Blood. 2012. PMID: 22645179 Free PMC article.
-
The very 5' end and the constant region of Ig genes are spared from somatic mutation because AID does not access these regions.J Exp Med. 2005 Nov 21;202(10):1443-54. doi: 10.1084/jem.20051604. J Exp Med. 2005. PMID: 16301749 Free PMC article.
-
Vpr expression abolishes the capacity of HIV-1 infected cells to repair uracilated DNA.Nucleic Acids Res. 2014 Feb;42(3):1698-710. doi: 10.1093/nar/gkt974. Epub 2013 Oct 30. Nucleic Acids Res. 2014. PMID: 24178031 Free PMC article.
-
Uracil DNA N-glycosylase promotes assembly of human centromere protein A.PLoS One. 2011 Mar 2;6(3):e17151. doi: 10.1371/journal.pone.0017151. PLoS One. 2011. PMID: 21399697 Free PMC article.
-
Replication Protein A Enhances Kinetics of Uracil DNA Glycosylase on ssDNA and Across DNA Junctions: Explored with a DNA Repair Complex Produced with SpyCatcher/SpyTag Ligation.Chembiochem. 2023 May 16;24(10):e202200765. doi: 10.1002/cbic.202200765. Epub 2023 Apr 18. Chembiochem. 2023. PMID: 36883884 Free PMC article.
References
-
- Krokan, H.E., H. Nilsen, F. Skorpen, M. Otterlei, and G. Slupphaug. 2000. Base excision repair of DNA in mammalian cells. FEBS Lett. 476:73–77. - PubMed
-
- Muramatsu, M., K. Kinoshita, S. Fagarasan, S. Yamada, Y. Shinkai, and T. Honjo. 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 102:553–563. - PubMed
-
- Revy, P., T. Muto, Y. Levy, F. Geissmann, A. Plebani, O. Sanal, N. Catalan, M. Forveille, R. Dufourcq-Labelouse, A. Gennery, et al. 2000. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell. 102:565–575. - PubMed
-
- Honjo, T., M. Muramatsu, and S. Fagarasan. 2004. AID: how does it aid antibody diversity? Immunity. 20:659–668. - PubMed
-
- Muramatsu, M., V.S. Sankaranand, S. Anant, M. Sugai, K. Kinoshita, N.O. Davidson, and T. Honjo. 1999. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J. Biol. Chem. 274:18470–18476. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
