

The nucleolus as a stress sensor: JNK2 inactivates the transcription factor TIF-IA and down-regulates rRNA synthesis
- PMID: 15805466
- PMCID: PMC1080132
- DOI: 10.1101/gad.333205
The nucleolus as a stress sensor: JNK2 inactivates the transcription factor TIF-IA and down-regulates rRNA synthesis
Abstract
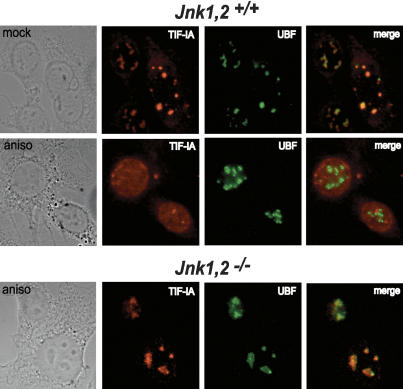
Cells respond to a variety of extracellular and intracellular forms of stress by down-regulating rRNA synthesis. We have investigated the mechanism underlying stress-dependent inhibition of RNA polymerase I (Pol I) transcription and show that the Pol I-specific transcription factor TIF-IA is inactivated upon stress. Inactivation is due to phosphorylation of TIF-IA by c-Jun N-terminal kinase (JNK) at a single threonine residue (Thr 200). Phosphorylation at Thr 200 impairs the interaction of TIF-IA with Pol I and the TBP-containing factor TIF-IB/SL1, thereby abrogating initiation complex formation. Moreover, TIF-IA is translocated from the nucleolus into the nucleoplasm. Substitution of Thr 200 by valine as well as knock-out of Jnk2 prevent inactivation and translocation of TIF-IA, leading to stress-resistance of Pol I transcription. Our data identify TIF-IA as a downstream target of the JNK pathway and suggest a critical role of JNK2 to protect rRNA synthesis against the harmful consequences of cellular stress.
Figures





Similar articles
-
Dynamic subcellular partitioning of the nucleolar transcription factor TIF-IA under ribotoxic stress.Biochim Biophys Acta. 2009 Jul;1793(7):1191-8. doi: 10.1016/j.bbamcr.2009.05.004. Epub 2009 May 18. Biochim Biophys Acta. 2009. PMID: 19450626
-
mTOR-dependent activation of the transcription factor TIF-IA links rRNA synthesis to nutrient availability.Genes Dev. 2004 Feb 15;18(4):423-34. doi: 10.1101/gad.285504. Genes Dev. 2004. PMID: 15004009 Free PMC article.
-
Akt activation enhances ribosomal RNA synthesis through casein kinase II and TIF-IA.Proc Natl Acad Sci U S A. 2013 Dec 17;110(51):20681-6. doi: 10.1073/pnas.1313097110. Epub 2013 Dec 2. Proc Natl Acad Sci U S A. 2013. PMID: 24297901 Free PMC article.
-
Cellular stress and nucleolar function.Cell Cycle. 2005 Aug;4(8):1036-8. doi: 10.4161/cc.4.8.1925. Epub 2005 Aug 20. Cell Cycle. 2005. PMID: 16205120 Review.
-
TIF-IA: An oncogenic target of pre-ribosomal RNA synthesis.Biochim Biophys Acta. 2016 Dec;1866(2):189-196. doi: 10.1016/j.bbcan.2016.09.003. Epub 2016 Sep 15. Biochim Biophys Acta. 2016. PMID: 27641688 Free PMC article. Review.
Cited by
-
Nucleolar stress in Drosophila melanogaster: RNAi-mediated depletion of Nopp140.Nucleus. 2013 Mar-Apr;4(2):123-33. doi: 10.4161/nucl.23944. Epub 2013 Feb 14. Nucleus. 2013. PMID: 23412656 Free PMC article.
-
Changes in DNA methylation patterns and repetitive sequences in blood lymphocytes of aged horses.Age (Dordr). 2014 Feb;36(1):31-48. doi: 10.1007/s11357-013-9541-z. Epub 2013 May 23. Age (Dordr). 2014. PMID: 23700175 Free PMC article.
-
Nucleolar AATF regulates c-Jun-mediated apoptosis.Mol Biol Cell. 2012 Nov;23(21):4323-32. doi: 10.1091/mbc.E12-05-0419. Epub 2012 Aug 29. Mol Biol Cell. 2012. PMID: 22933572 Free PMC article.
-
TOR-dependent reduction in the expression level of Rrn3p lowers the activity of the yeast RNA Pol I machinery, but does not account for the strong inhibition of rRNA production.Nucleic Acids Res. 2010 Sep;38(16):5315-26. doi: 10.1093/nar/gkq264. Epub 2010 Apr 25. Nucleic Acids Res. 2010. PMID: 20421203 Free PMC article.
-
STAT3 potentiates RNA polymerase I-directed transcription and tumor growth by activating RPA34 expression.Br J Cancer. 2023 Mar;128(5):766-782. doi: 10.1038/s41416-022-02098-6. Epub 2022 Dec 16. Br J Cancer. 2023. PMID: 36526675 Free PMC article.
References
-
- Barr R.K. and Bogoyevitch, M.A. 2001. The c-jun N-terminal kinase family of mitogen-activated protein kinases (JNK MAPKs). Int. J. Biochem. Cell Biol. 33: 1047-1063. - PubMed
-
- Boyle W.J., van der Geer, P., and Hunter, T. 1991. Phosphopeptide mapping and phosphoamino acid analysis by two-dimensional separation on thin-layer cellulose plates. Meth. Enzymol. 201: 110-149. - PubMed
-
- Cavanaugh A.H., Hirschler-Laszkiewicz, I., Hu, Q., Dundr, M., Smink, T., Misteli, T., and Rothblum, L.I. 2002. Rrn3 phosphorylation is a regulatory checkpoint for ribosome biogenesis. J. Biol. Chem. 277: 27423-27432. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous