

Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF
- PMID: 15710891
- PMCID: PMC548795
- DOI: 10.1073/pnas.0408824102
Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF
Abstract
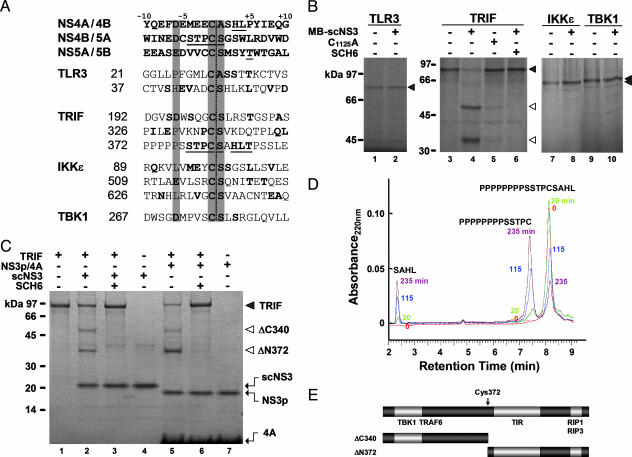
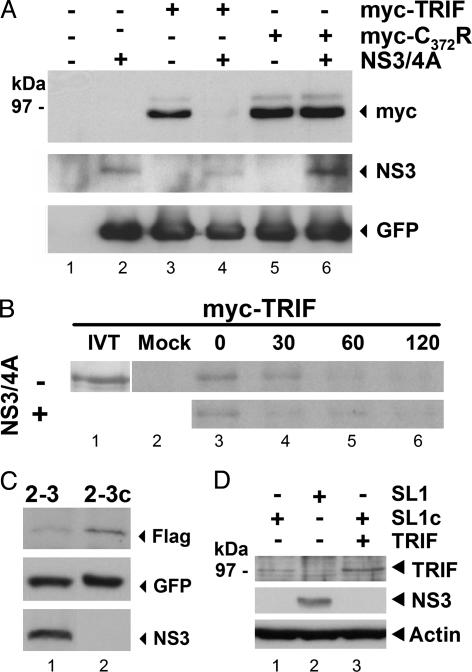
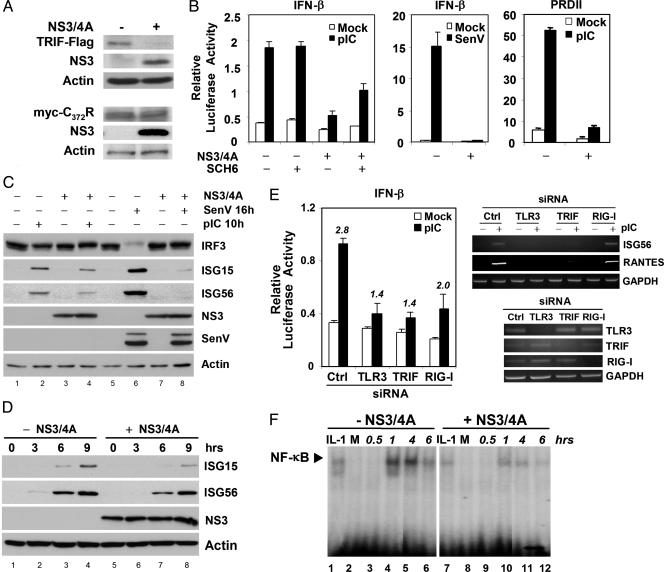
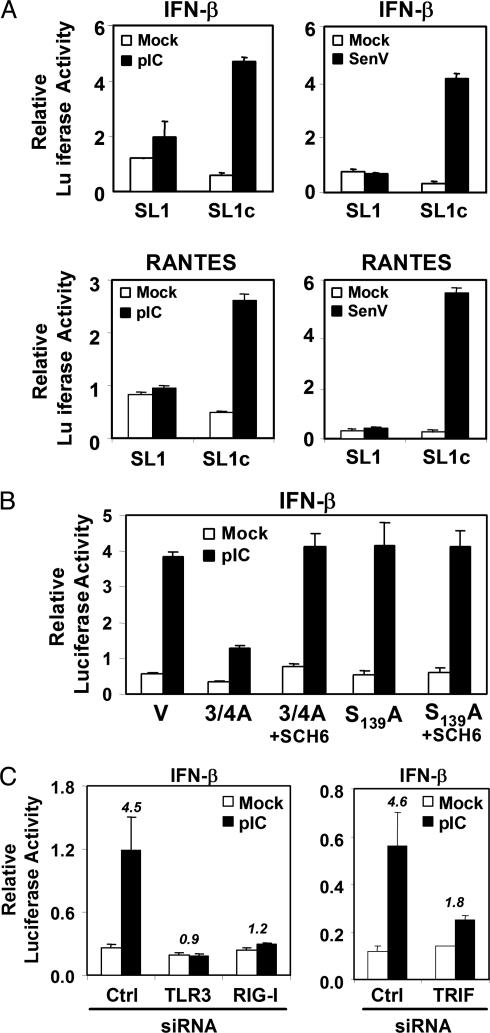
Toll-like receptors (TLRs) bind pathogen-specific ligands early in infection, initiating signaling pathways that lead to expression of multiple protective cellular genes. Many viruses have evolved strategies that block the effector mechanisms induced through these signaling pathways, but viral interference with critical proximal receptor interactions has not been described. We show here that the NS3/4A serine protease of hepatitis C virus (HCV), a virus notorious for its ability to establish persistent intrahepatic infection, causes specific proteolysis of Toll-IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF or TICAM-1), an adaptor protein linking TLR3 to kinases responsible for activating IFN regulatory factor 3 (IRF-3) and NF-kappaB, transcription factors controlling a multiplicity of antiviral defenses. NS3/4A-mediated cleavage of TRIF reduces its abundance and inhibits polyI:C-activated signaling through the TLR3 pathway before its bifurcation to IRF-3 and NF-kappaB. This uniquely broad mechanism of immune evasion potentially limits expression of multiple host defense genes, thereby promoting persistent infections with this medically important virus.
Figures
Similar articles
-
Toll-like receptor 3-mediated activation of NF-kappaB and IRF3 diverges at Toll-IL-1 receptor domain-containing adapter inducing IFN-beta.Proc Natl Acad Sci U S A. 2004 Mar 9;101(10):3533-8. doi: 10.1073/pnas.0308496101. Epub 2004 Feb 24. Proc Natl Acad Sci U S A. 2004. PMID: 14982987 Free PMC article.
-
Inhibition of RIG-I-dependent signaling to the interferon pathway during hepatitis C virus expression and restoration of signaling by IKKepsilon.J Virol. 2005 Apr;79(7):3969-78. doi: 10.1128/JVI.79.7.3969-3978.2005. J Virol. 2005. PMID: 15767399 Free PMC article.
-
Limited suppression of the interferon-beta production by hepatitis C virus serine protease in cultured human hepatocytes.FEBS J. 2007 Aug;274(16):4161-76. doi: 10.1111/j.1742-4658.2007.05942.x. Epub 2007 Jul 25. FEBS J. 2007. PMID: 17651439
-
[Toll-like receptors that sense viral infection].Uirusu. 2004 Jun;54(1):1-8. doi: 10.2222/jsv.54.1. Uirusu. 2004. PMID: 15449898 Review. Japanese.
-
TICAM-1 and TICAM-2: toll-like receptor adapters that participate in induction of type 1 interferons.Int J Biochem Cell Biol. 2005 Mar;37(3):524-9. doi: 10.1016/j.biocel.2004.07.018. Int J Biochem Cell Biol. 2005. PMID: 15618008 Review.
Cited by
-
dsRNA-dependent protein kinase PKR and its role in stress, signaling and HCV infection.Viruses. 2012 Oct 29;4(11):2598-635. doi: 10.3390/v4112598. Viruses. 2012. PMID: 23202496 Free PMC article. Review.
-
An Internally Translated MAVS Variant Exposes Its Amino-terminal TRAF-Binding Motifs to Deregulate Interferon Induction.PLoS Pathog. 2015 Jul 29;11(7):e1005060. doi: 10.1371/journal.ppat.1005060. eCollection 2015 Jul. PLoS Pathog. 2015. PMID: 26221961 Free PMC article.
-
Regulation of hepatitis C virus replication and gene expression by the MAPK-ERK pathway.Virol Sin. 2012 Oct;27(5):278-85. doi: 10.1007/s12250-012-3257-6. Epub 2012 Sep 21. Virol Sin. 2012. PMID: 23001481 Free PMC article.
-
Integrated analysis to study the interplay between post-translational modifications (PTM) in hepatitis C virus proteins and hepatocellular carcinoma (HCC) development.Sci Rep. 2022 Sep 19;12(1):15648. doi: 10.1038/s41598-022-19854-6. Sci Rep. 2022. PMID: 36123370 Free PMC article.
-
Toll-like receptors in liver fibrosis: cellular crosstalk and mechanisms.Front Physiol. 2012 May 22;3:138. doi: 10.3389/fphys.2012.00138. eCollection 2012. Front Physiol. 2012. PMID: 22661952 Free PMC article.
References
-
- Seeff, L. B. (1997) Hepatology 26, 21S-28S. - PubMed
-
- Racanelli, V. & Rehermann, B. (2003) Trends Immunol. 24, 456-464. - PubMed
-
- Foy, E., Li, K., Wang, C., Sumter, R., Ikeda, M., Lemon, S. M. & Gale, M., Jr. (2003) Science 300, 1145-1148. - PubMed
-
- Hiscott, J., Pitha, P., Genin, P., Nguyen, H., Heylbroeck, C., Mamane, Y., Algarte, M. & Lin, R. (1999) J. Interferon Cytokine Res. 19, 1-13. - PubMed
-
- Yoneyama, M., Suhara, W. & Fujita, T. (2002) J. Interferon Cytokine Res. 22, 73-76. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
