

Liver fibrosis
- PMID: 15690074
- PMCID: PMC546435
- DOI: 10.1172/JCI24282
Liver fibrosis
Erratum in
- J Clin Invest. 2005 Apr;115(4):1100
Abstract
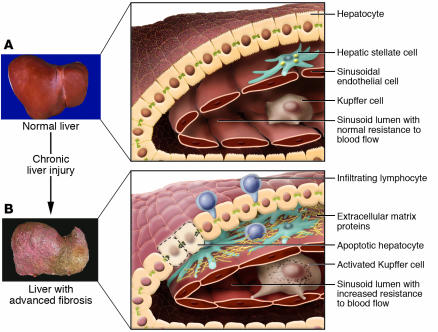
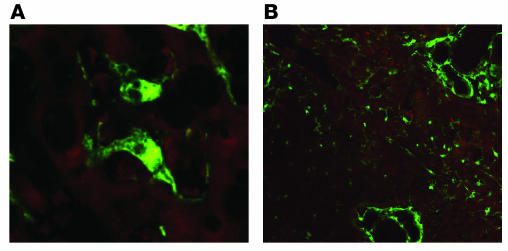
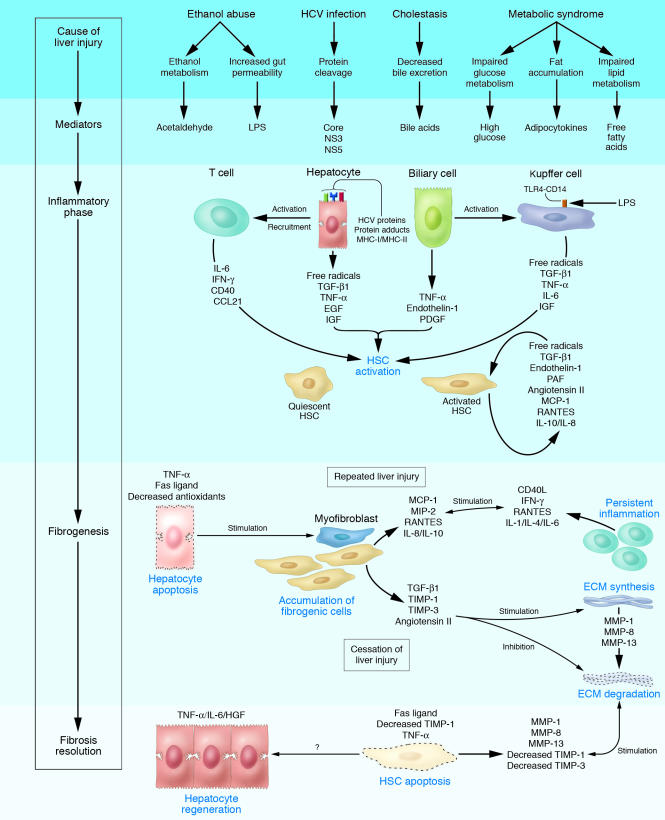
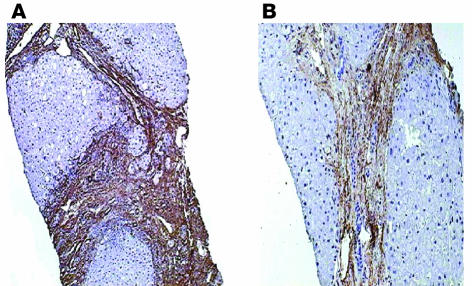
Liver fibrosis is the excessive accumulation of extracellular matrix proteins including collagen that occurs in most types of chronic liver diseases. Advanced liver fibrosis results in cirrhosis, liver failure, and portal hypertension and often requires liver transplantation. Our knowledge of the cellular and molecular mechanisms of liver fibrosis has greatly advanced. Activated hepatic stellate cells, portal fibroblasts, and myofibroblasts of bone marrow origin have been identified as major collagen-producing cells in the injured liver. These cells are activated by fibrogenic cytokines such as TGF-beta1, angiotensin II, and leptin. Reversibility of advanced liver fibrosis in patients has been recently documented, which has stimulated researchers to develop antifibrotic drugs. Emerging antifibrotic therapies are aimed at inhibiting the accumulation of fibrogenic cells and/or preventing the deposition of extracellular matrix proteins. Although many therapeutic interventions are effective in experimental models of liver fibrosis, their efficacy and safety in humans is unknown. This review summarizes recent progress in the study of the pathogenesis and diagnosis of liver fibrosis and discusses current antifibrotic strategies.
Figures
Similar articles
-
Graptopetalum paraguayense Inhibits Liver Fibrosis by Blocking TGF-β Signaling In Vivo and In Vitro.Int J Mol Sci. 2019 May 27;20(10):2592. doi: 10.3390/ijms20102592. Int J Mol Sci. 2019. PMID: 31137784 Free PMC article.
-
Hepatic stellate cells as a target for the treatment of liver fibrosis.Semin Liver Dis. 2001 Aug;21(3):437-51. doi: 10.1055/s-2001-17558. Semin Liver Dis. 2001. PMID: 11586471 Review.
-
Anti-fibrogenic strategies and the regression of fibrosis.Best Pract Res Clin Gastroenterol. 2011 Apr;25(2):305-17. doi: 10.1016/j.bpg.2011.02.011. Best Pract Res Clin Gastroenterol. 2011. PMID: 21497747 Free PMC article. Review.
-
Liver fibrosis: Direct antifibrotic agents and targeted therapies.Matrix Biol. 2018 Aug;68-69:435-451. doi: 10.1016/j.matbio.2018.04.006. Epub 2018 Apr 12. Matrix Biol. 2018. PMID: 29656147 Review.
-
Relaxin in hepatic fibrosis: What is known and where to head?Biochimie. 2021 Aug;187:144-151. doi: 10.1016/j.biochi.2021.06.001. Epub 2021 Jun 5. Biochimie. 2021. PMID: 34102254 Review.
Cited by
-
Pathophysiology and Management of Variceal Bleeding.Drugs. 2021 Apr;81(6):647-667. doi: 10.1007/s40265-021-01493-2. Epub 2021 Mar 12. Drugs. 2021. PMID: 33710585 Review.
-
Development of Adenoviral Delivery Systems to Target Hepatic Stellate Cells In Vivo.PLoS One. 2013 Jun 18;8(6):e67091. doi: 10.1371/journal.pone.0067091. Print 2013. PLoS One. 2013. PMID: 23825626 Free PMC article.
-
Hepatic stellate cells--the pericytes in the liver.Pflugers Arch. 2013 Jun;465(6):775-8. doi: 10.1007/s00424-012-1209-5. Epub 2013 Jan 5. Pflugers Arch. 2013. PMID: 23292551 Review.
-
Mesenchymal Stem/Stromal Cells in Liver Fibrosis: Recent Findings, Old/New Caveats and Future Perspectives.Stem Cell Rev Rep. 2015 Aug;11(4):586-97. doi: 10.1007/s12015-015-9585-9. Stem Cell Rev Rep. 2015. PMID: 25820543 Review.
-
Intra-individual comparison of liver stiffness measurements by magnetic resonance elastography and two-dimensional shear-wave elastography in 888 patients.Ultrasonography. 2023 Jan;42(1):65-77. doi: 10.14366/usg.22052. Epub 2022 Jun 21. Ultrasonography. 2023. PMID: 36366945 Free PMC article.
References
-
- Friedman SL. Liver fibrosis - from bench to bedside. J. Hepatol. 2003;38(Suppl. 1):S38–S53. - PubMed
-
- Gines P, Cardenas A, Arroyo V, Rodes J. Management of cirrhosis and ascites. N. Engl. J. Med. 2004;350:1646–1654. - PubMed
-
- Popper H, Uenfriend S. Hepatic fibrosis. Correlation of biochemical and morphologic investigations. Am. J. Med. 1970;49:707–721. - PubMed
-
- Schaffner F, Klion FM. Chronic hepatitis. Annu. Rev. Med. 1968;19:25–38. - PubMed
-
- Albanis E, Friedman SL. Hepatic fibrosis. Pathogenesis and principles of therapy. Clin. Liver Dis. 2001;5:315–334, v–vi. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
