

Complete genome sequence of the probiotic lactic acid bacterium Lactobacillus acidophilus NCFM
- PMID: 15671160
- PMCID: PMC554803
- DOI: 10.1073/pnas.0409188102
Complete genome sequence of the probiotic lactic acid bacterium Lactobacillus acidophilus NCFM
Abstract
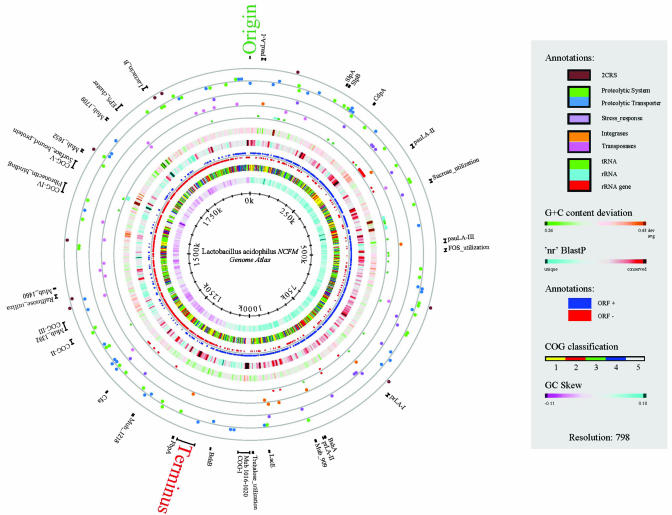
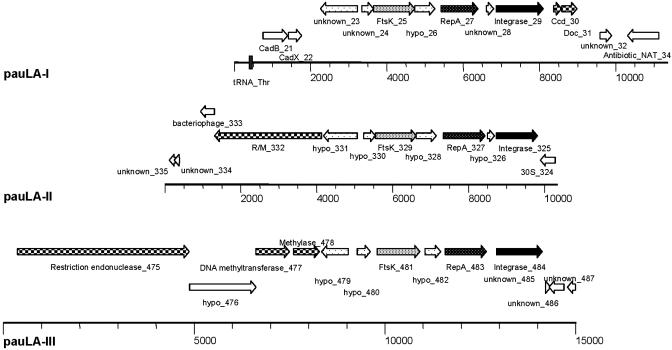
Lactobacillus acidophilus NCFM is a probiotic bacterium that has been produced commercially since 1972. The complete genome is 1,993,564 nt and devoid of plasmids. The average GC content is 34.71% with 1,864 predicted ORFs, of which 72.5% were functionally classified. Nine phage-related integrases were predicted, but no complete prophages were found. However, three unique regions designated as potential autonomous units (PAUs) were identified. These units resemble a unique structure and bear characteristics of both plasmids and phages. Analysis of the three PAUs revealed the presence of two R/M systems and a prophage maintenance system killer protein. A spacers interspersed direct repeat locus containing 32 nearly perfect 29-bp repeats was discovered and may provide a unique molecular signature for this organism. In silico analyses predicted 17 transposase genes and a chromosomal locus for lactacin B, a class II bacteriocin. Several mucus- and fibronectin-binding proteins, implicated in adhesion to human intestinal cells, were also identified. Gene clusters for transport of a diverse group of carbohydrates, including fructooligosaccharides and raffinose, were present and often accompanied by transcriptional regulators of the lacI family. For protein degradation and peptide utilization, the organism encoded 20 putative peptidases, homologs for PrtP and PrtM, and two complete oligopeptide transport systems. Nine two-component regulatory systems were predicted, some associated with determinants implicated in bacteriocin production and acid tolerance. Collectively, these features within the genome sequence of L. acidophilus are likely to contribute to the organisms' gastric survival and promote interactions with the intestinal mucosa and microbiota.
Figures

Similar articles
-
Temporal gene expression and probiotic attributes of Lactobacillus acidophilus during growth in milk.J Dairy Sci. 2009 Mar;92(3):870-86. doi: 10.3168/jds.2008-1457. J Dairy Sci. 2009. PMID: 19233780
-
Mucin- and carbohydrate-stimulated adhesion and subproteome changes of the probiotic bacterium Lactobacillus acidophilus NCFM.J Proteomics. 2017 Jun 23;163:102-110. doi: 10.1016/j.jprot.2017.05.015. Epub 2017 May 19. J Proteomics. 2017. PMID: 28533178
-
Identification of an operon and inducing peptide involved in the production of lactacin B by Lactobacillus acidophilus.J Appl Microbiol. 2007 Nov;103(5):1766-78. doi: 10.1111/j.1365-2672.2007.03417.x. J Appl Microbiol. 2007. PMID: 17953587
-
Low-redundancy sequencing of the entire Lactococcus lactis IL1403 genome.Antonie Van Leeuwenhoek. 1999 Jul-Nov;76(1-4):27-76. Antonie Van Leeuwenhoek. 1999. PMID: 10532372 Review.
-
Invited review: the scientific basis of Lactobacillus acidophilus NCFM functionality as a probiotic.J Dairy Sci. 2001 Feb;84(2):319-31. doi: 10.3168/jds.S0022-0302(01)74481-5. J Dairy Sci. 2001. PMID: 11233016 Review.
Cited by
-
An In Vitro Colonic Fermentation Study of the Effects of Human Milk Oligosaccharides on Gut Microbiota and Short-Chain Fatty Acid Production in Infants Aged 0-6 Months.Foods. 2024 Mar 18;13(6):921. doi: 10.3390/foods13060921. Foods. 2024. PMID: 38540911 Free PMC article.
-
Detection of oxalyl-CoA decarboxylase (oxc) and formyl-CoA transferase (frc) genes in novel probiotic isolates capable of oxalate degradation in vitro.Folia Microbiol (Praha). 2024 Apr;69(2):423-432. doi: 10.1007/s12223-024-01128-5. Epub 2024 Jan 13. Folia Microbiol (Praha). 2024. PMID: 38217756 Free PMC article.
-
Evaluation of Prebiotic Activity of Stellariae Radix Polysaccharides and Its Effects on Gut Microbiota.Nutrients. 2023 Nov 20;15(22):4843. doi: 10.3390/nu15224843. Nutrients. 2023. PMID: 38004237 Free PMC article.
-
In silico prospection of Lactobacillus acidophilus strains with potential probiotic activity.Braz J Microbiol. 2023 Dec;54(4):2733-2743. doi: 10.1007/s42770-023-01139-3. Epub 2023 Oct 6. Braz J Microbiol. 2023. PMID: 37801223 Free PMC article.
-
Molecular strategies for the utilisation of human milk oligosaccharides by infant gut-associated bacteria.FEMS Microbiol Rev. 2023 Nov 1;47(6):fuad056. doi: 10.1093/femsre/fuad056. FEMS Microbiol Rev. 2023. PMID: 37793834 Free PMC article. Review.
References
-
- Reid, G., Sanders, M. E., Gaskins, H. R., Gibson, G. R., Mercenier, A., Rastall, R., Roberfroid, M., Rowland, I., Cherbut, C. & Klaenhammer, T. R. (2003) J. Clin. Gastroenterol. 37, 105–118. - PubMed
-
- Klaenhammer, T. R. & Russell, W. M. (2000) in Encyclopedia of Food Microbiology (Academic, Amsterdam), Vol. 2, pp. 1151–1157.
-
- Johnson, J. L., Phelps, C. F., Cummins, C. S., London, J. & Gasser, F. (1980) Int. J. Syst. Bacteriol. 30, 53–68.
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
