

The Drosha-DGCR8 complex in primary microRNA processing
- PMID: 15574589
- PMCID: PMC535913
- DOI: 10.1101/gad.1262504
The Drosha-DGCR8 complex in primary microRNA processing
Abstract
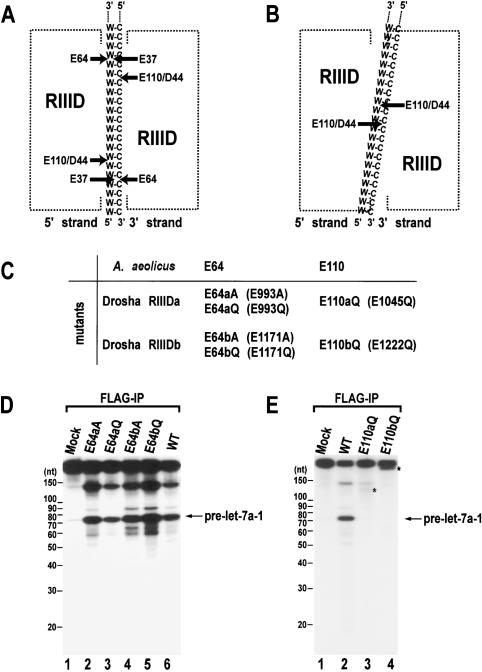
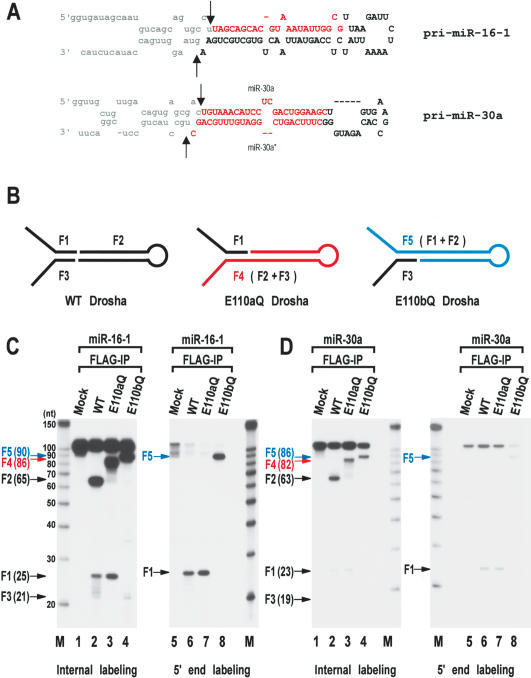
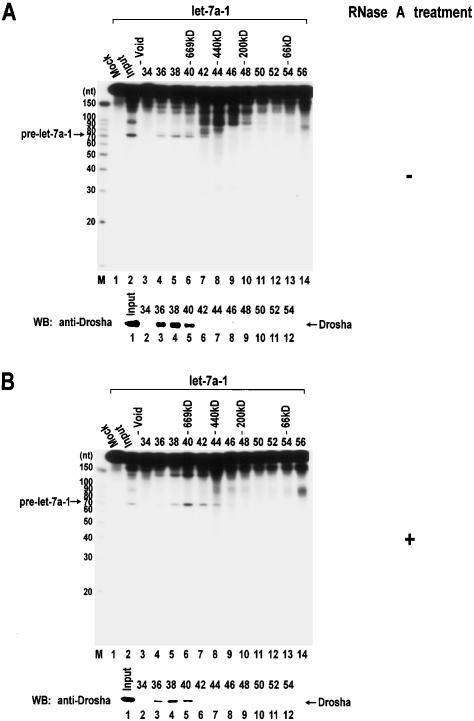
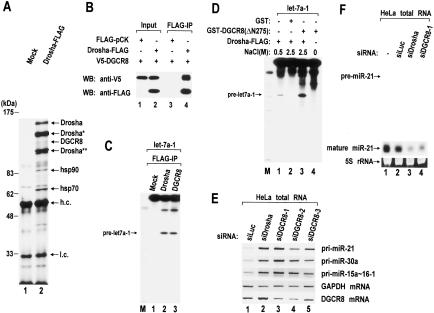
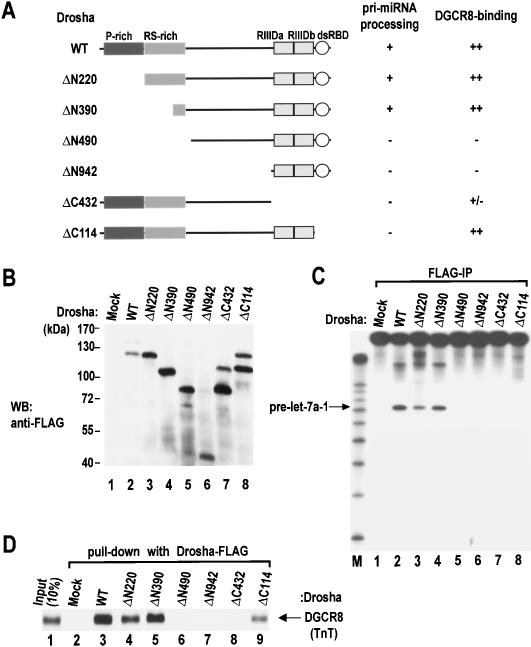
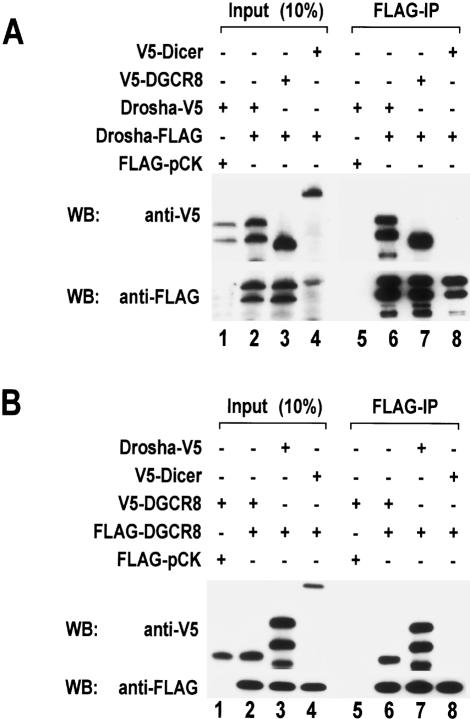
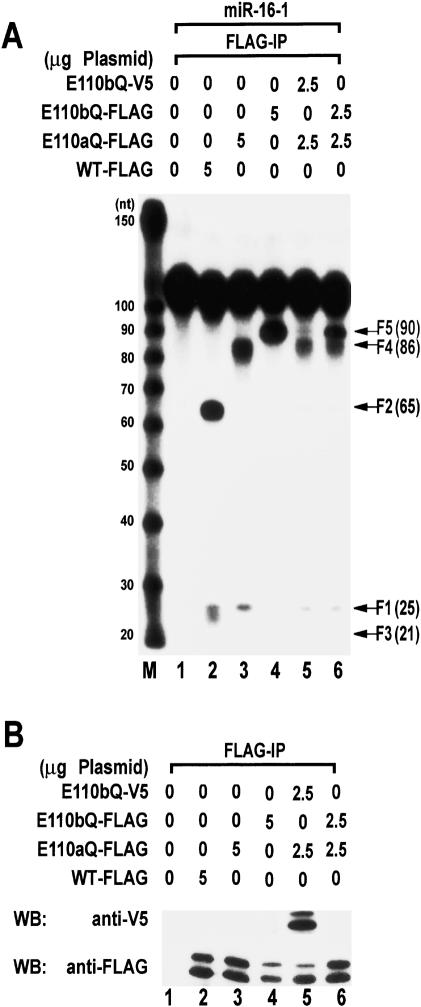
RNase III proteins play key roles in microRNA (miRNA) biogenesis. The nuclear RNase III Drosha cleaves primary miRNAs (pri-miRNAs) to release hairpin-shaped pre-miRNAs that are subsequently cut by the cytoplasmic RNase III Dicer to generate mature miRNAs. While Dicer (class III) and other simple RNase III proteins (class I) have been studied intensively, the class II enzyme Drosha remains to be characterized. Here we dissected the action mechanism of human Drosha by generating mutants and by characterizing its new interacting partner, DGCR8. The basic action mechanism of Drosha was found to be similar to that of human Dicer; the RNase III domains A and B form an intramolecular dimer and cleave the 3' and 5' strands of the stem, respectively. Human Drosha fractionates at approximately 650 kDa, indicating that Drosha functions as a large complex. In this complex, Drosha interacts with DGCR8, which contains two double-stranded RNA (dsRNA)-binding domains. By RNAi and biochemical reconstitution, we show that DGCR8 may be an essential component of the pri-miRNA processing complex, along with Drosha. Based on these results, we propose a model for the action mechanism of class II RNase III proteins.
Figures
Similar articles
-
Characterization of DGCR8/Pasha, the essential cofactor for Drosha in primary miRNA processing.Nucleic Acids Res. 2006;34(16):4622-9. doi: 10.1093/nar/gkl458. Epub 2006 Sep 8. Nucleic Acids Res. 2006. PMID: 16963499 Free PMC article.
-
Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex.Cell. 2006 Jun 2;125(5):887-901. doi: 10.1016/j.cell.2006.03.043. Cell. 2006. PMID: 16751099
-
MicroRNA biogenesis: isolation and characterization of the microprocessor complex.Methods Mol Biol. 2006;342:33-47. doi: 10.1385/1-59745-123-1:33. Methods Mol Biol. 2006. PMID: 16957365 Review.
-
In vitro and in vivo assays for the activity of Drosha complex.Methods Enzymol. 2007;427:89-106. doi: 10.1016/S0076-6879(07)27005-3. Methods Enzymol. 2007. PMID: 17720480
-
Rethinking the microprocessor.Cell. 2006 Jun 2;125(5):827-9. doi: 10.1016/j.cell.2006.05.018. Cell. 2006. PMID: 16751089 Review.
Cited by
-
The development and controversy of competitive endogenous RNA hypothesis in non-coding genes.Mol Cell Biochem. 2021 Jan;476(1):109-123. doi: 10.1007/s11010-020-03889-2. Epub 2020 Sep 25. Mol Cell Biochem. 2021. PMID: 32975695 Review.
-
The kinase ABL phosphorylates the microprocessor subunit DGCR8 to stimulate primary microRNA processing in response to DNA damage.Sci Signal. 2015 Jun 30;8(383):ra64. doi: 10.1126/scisignal.aaa4468. Sci Signal. 2015. PMID: 26126715 Free PMC article.
-
Identification of Dirofilaria immitis miRNA using illumina deep sequencing.Vet Res. 2013 Jan 18;44(1):3. doi: 10.1186/1297-9716-44-3. Vet Res. 2013. PMID: 23331513 Free PMC article.
-
PKM2 promotes glucose metabolism and cell growth in gliomas through a mechanism involving a let-7a/c-Myc/hnRNPA1 feedback loop.Oncotarget. 2015 May 30;6(15):13006-18. doi: 10.18632/oncotarget.3514. Oncotarget. 2015. PMID: 25948776 Free PMC article.
-
miR-196, an Emerging Cancer Biomarker for Digestive Tract Cancers.J Cancer. 2016 Mar 20;7(6):650-5. doi: 10.7150/jca.13460. eCollection 2016. J Cancer. 2016. PMID: 27076845 Free PMC article. Review.
References
-
- Bartel D.P. 2004. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 116: 281-297. - PubMed
-
- Bernstein E., Caudy, A.A., Hammond, S.M., and Hannon, G.J. 2001. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409: 363-366. - PubMed
-
- Blaszczyk J., Tropea, J.E., Bubunenko, M., Routzahn, K.M., Waugh, D.S., Court, D.L., and Ji, X. 2001. Crystallographic and modeling studies of RNase III suggest a mechanism for double-stranded RNA cleavage. Structure (Camb) 9: 1225-1236. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases