

Molecular, biological, and in vivo characterization of the guinea pig cytomegalovirus (CMV) homologs of the human CMV matrix proteins pp71 (UL82) and pp65 (UL83)
- PMID: 15331722
- PMCID: PMC515002
- DOI: 10.1128/JVI.78.18.9872-9889.2004
Molecular, biological, and in vivo characterization of the guinea pig cytomegalovirus (CMV) homologs of the human CMV matrix proteins pp71 (UL82) and pp65 (UL83)
Abstract
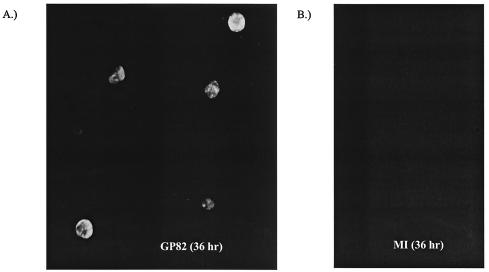
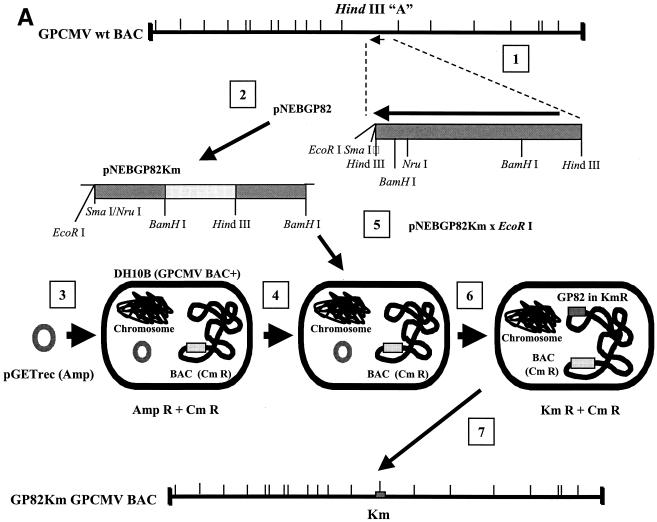
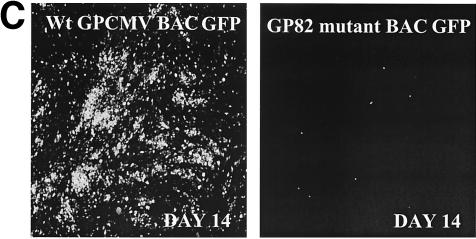
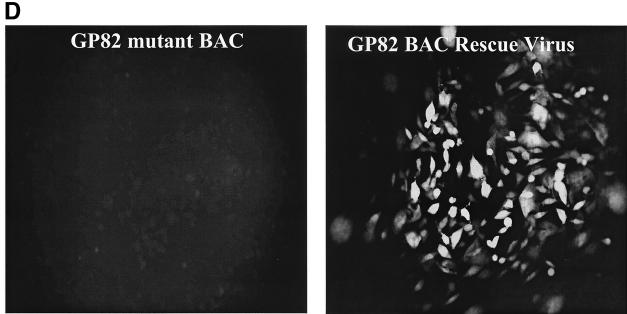
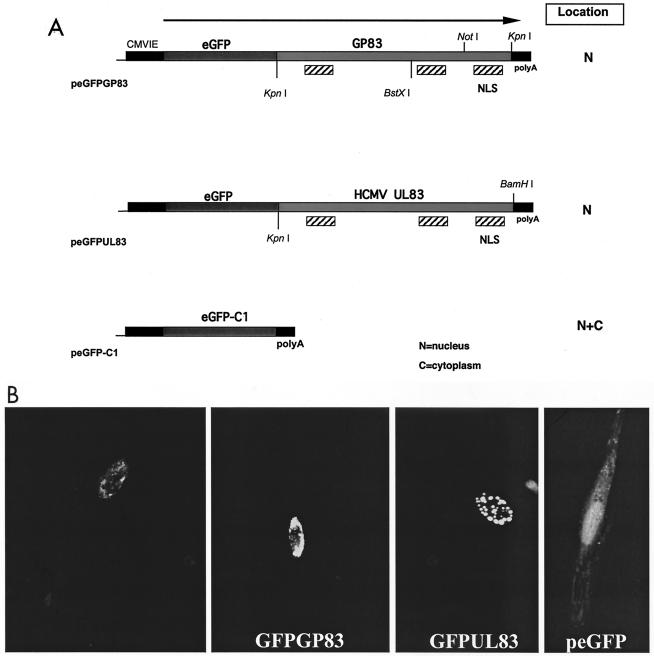
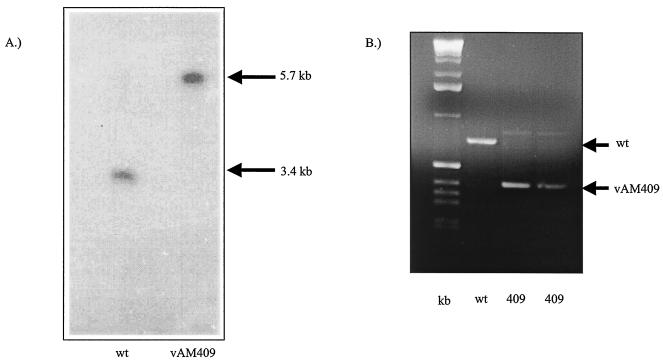
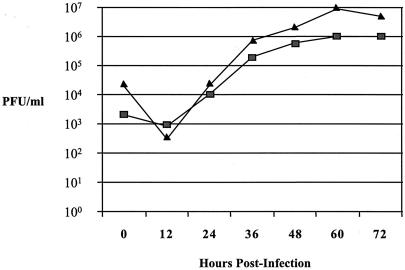
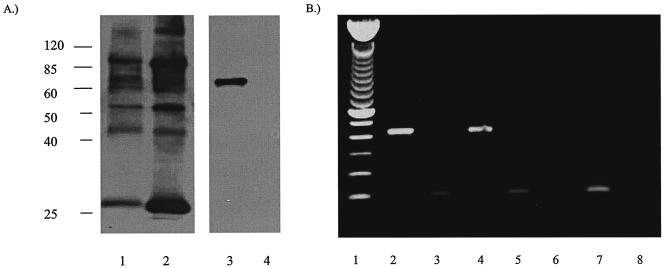
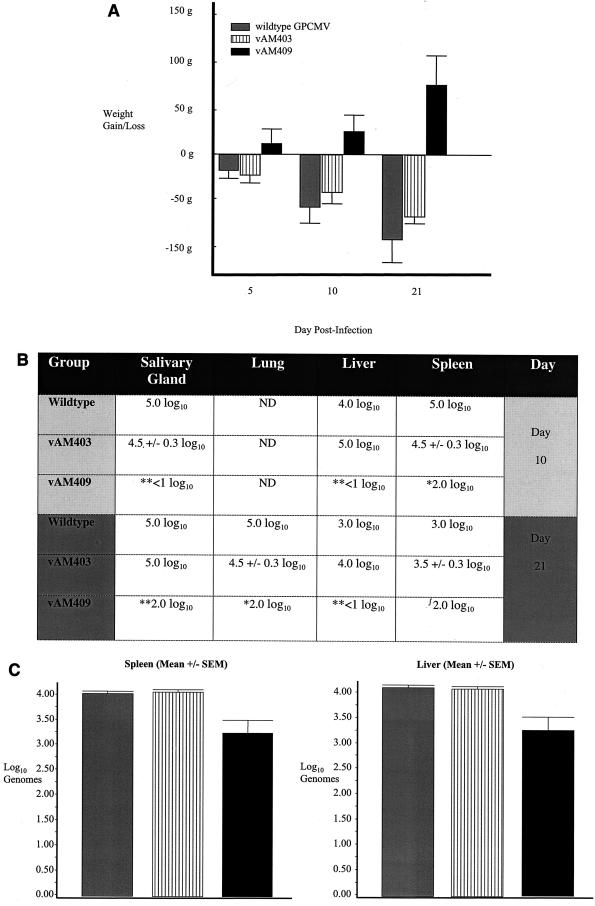
We recently identified the genes encoding the guinea pig cytomegalovirus (GPCMV) homologs of the upper and lower matrix proteins of human CMV, pp71 (UL82) and pp65 (UL83), which we designated GP82 and GP83, respectively. Transient-expression studies with a GP82 plasmid demonstrated that the encoded protein targets the nucleus and that the infectivity and plaquing efficiency of cotransfected GPCMV viral DNA was enhanced by GP82. The transactivation function of GP82 was not limited to GPCMV, but was also observed for a heterologous virus, herpes simplex virus type 1 (HSV-1). This was confirmed by its ability to complement the growth of an HSV-1 VP16 transactivation-defective mutant virus in an HSV viral DNA cotransfection assay. Study of a GP82 "knockout" virus (and its attendant rescuant), generated on a GPCMV bacterial artificial chromosome construct, confirmed the essential nature of the gene. Conventional homologous recombination was used to generate a GP83 mutant to examine the role of GP83 in the viral life cycle. Comparison of the one-step growth kinetics of the GP83 mutant (vAM409) and wild-type GPCMV indicated that GP83 protein is not required for viral replication in tissue culture. The role of GP83 in vivo was examined by comparing the pathogenesis of wild-type GPCMV, vAM409, and a control virus, vAM403, in guinea pigs. The vAM409 mutant was significantly attenuated for dissemination in immunocompromised strain 2 guinea pigs, suggesting that the GP83 protein is essential for full pathogenicity in vivo.
Copyright 2004 American Society for Microbiology
Figures





Similar articles
-
A Fully Protective Congenital CMV Vaccine Requires Neutralizing Antibodies to Viral Pentamer and gB Glycoprotein Complexes but a pp65 T-Cell Response Is Not Necessary.Viruses. 2021 Jul 27;13(8):1467. doi: 10.3390/v13081467. Viruses. 2021. PMID: 34452332 Free PMC article.
-
Molecular cloning of the guinea pig cytomegalovirus (GPCMV) genome as an infectious bacterial artificial chromosome (BAC) in Escherichia coli.Mol Genet Metab. 2001 Jan;72(1):15-26. doi: 10.1006/mgme.2000.3102. Mol Genet Metab. 2001. PMID: 11161824
-
Preconceptual administration of an alphavirus replicon UL83 (pp65 homolog) vaccine induces humoral and cellular immunity and improves pregnancy outcome in the guinea pig model of congenital cytomegalovirus infection.J Infect Dis. 2007 Mar 15;195(6):789-98. doi: 10.1086/511982. Epub 2007 Feb 6. J Infect Dis. 2007. PMID: 17299708
-
Animal models of congenital cytomegalovirus infection: an overview of progress in the characterization of guinea pig cytomegalovirus (GPCMV).J Clin Virol. 2002 Aug;25 Suppl 2:S37-49. doi: 10.1016/s1386-6532(02)00100-2. J Clin Virol. 2002. PMID: 12361755 Review.
-
Comparison of vaccine strategies against congenital CMV infection in the guinea pig model.J Clin Virol. 2008 Mar;41(3):224-30. doi: 10.1016/j.jcv.2007.10.008. Epub 2007 Dec 3. J Clin Virol. 2008. PMID: 18060834 Review.
Cited by
-
Enhancing safety of cytomegalovirus-based vaccine vectors by engaging host intrinsic immunity.Sci Transl Med. 2019 Jul 17;11(501):eaaw2603. doi: 10.1126/scitranslmed.aaw2603. Sci Transl Med. 2019. PMID: 31316006 Free PMC article.
-
Guinea pig cytomegalovirus GP84 is a functional homolog of the human cytomegalovirus (HCMV) UL84 gene that can complement for the loss of UL84 in a chimeric HCMV.Virology. 2011 Feb 5;410(1):76-87. doi: 10.1016/j.virol.2010.10.028. Epub 2010 Nov 20. Virology. 2011. PMID: 21094510 Free PMC article.
-
Cytomegalovirus pp65 limits dissemination but is dispensable for persistence.J Clin Invest. 2014 May;124(5):1928-44. doi: 10.1172/JCI67420. Epub 2014 Apr 1. J Clin Invest. 2014. PMID: 24691437 Free PMC article.
-
Virion factors that target Daxx to overcome intrinsic immunity.J Virol. 2013 Oct;87(19):10412-22. doi: 10.1128/JVI.00425-13. Epub 2013 Jul 17. J Virol. 2013. PMID: 23864634 Free PMC article. Review.
-
Inclusion of the Viral Pentamer Complex in a Vaccine Design Greatly Improves Protection against Congenital Cytomegalovirus in the Guinea Pig Model.J Virol. 2019 Oct 29;93(22):e01442-19. doi: 10.1128/JVI.01442-19. Print 2019 Nov 15. J Virol. 2019. PMID: 31484753 Free PMC article.
References
-
- Berencsi, K., Z. Gyulai, E. Gonczol, S. Pincus, W. I. Cox, S. Michelson, L. Kari, C. Meric, M. Cadoz, J. Zahradnik, S. Starr, and S. Plotkin. 2001. A canarypox vector-expressing cytomegalovirus (CMV) phosphoprotein 65 induces long-lasting cytotoxic T cell responses in human CMV-seronegative subjects. J. Infect. Dis. 183:1171-1179. - PubMed
-
- Bia, F. J., B. P. Griffith, C. K. Fong, and G. D. Hsiung. 1983. Cytomegaloviral infections in the guinea pig: experimental models for human disease. Rev. Infect. Dis. 5:177-195. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
