

Cancer cachexia is regulated by selective targeting of skeletal muscle gene products
- PMID: 15286803
- PMCID: PMC484974
- DOI: 10.1172/JCI20174
Cancer cachexia is regulated by selective targeting of skeletal muscle gene products
Abstract
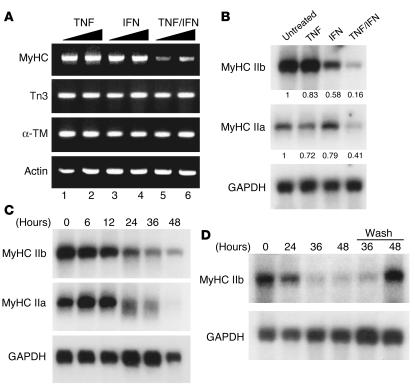
Cachexia is a syndrome characterized by wasting of skeletal muscle and contributes to nearly one-third of all cancer deaths. Cytokines and tumor factors mediate wasting by suppressing muscle gene products, but exactly which products are targeted by these cachectic factors is not well understood. Because of their functional relevance to muscle architecture, such targets are presumed to represent myofibrillar proteins, but whether these proteins are regulated in a general or a selective manner is also unclear. Here we demonstrate, using in vitro and in vivo models of muscle wasting, that cachectic factors are remarkably selective in targeting myosin heavy chain. In myotubes and mouse muscles, TNF-alpha plus IFN-gamma strongly reduced myosin expression through an RNA-dependent mechanism. Likewise, colon-26 tumors in mice caused the selective reduction of this myofibrillar protein, and this reduction correlated with wasting. Under these conditions, however, loss of myosin was associated with the ubiquitin-dependent proteasome pathway, which suggests that mechanisms used to regulate the expression of muscle proteins may be cachectic factor specific. These results shed new light on cancer cachexia by revealing that wasting does not result from a general downregulation of muscle proteins but rather is highly selective as to which proteins are targeted during the wasting state.
Figures





Similar articles
-
NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia.Science. 2000 Sep 29;289(5488):2363-6. doi: 10.1126/science.289.5488.2363. Science. 2000. PMID: 11009425
-
Biomedicine. Protein loss in cancer cachexia.Science. 2000 Sep 29;289(5488):2293-4. doi: 10.1126/science.289.5488.2293. Science. 2000. PMID: 11041796 No abstract available.
-
Cachexia in cancer--zeroing in on myosin.N Engl J Med. 2004 Nov 11;351(20):2124-5. doi: 10.1056/NEJMcibr042889. N Engl J Med. 2004. PMID: 15537911 No abstract available.
-
Adaptation of the ubiquitin-proteasome proteolytic pathway in cancer cachexia.Mol Biol Rep. 1999 Apr;26(1-2):77-82. doi: 10.1023/a:1006961919775. Mol Biol Rep. 1999. PMID: 10363651 Review.
-
TNF-α and cancer cachexia: Molecular insights and clinical implications.Life Sci. 2017 Feb 1;170:56-63. doi: 10.1016/j.lfs.2016.11.033. Epub 2016 Dec 3. Life Sci. 2017. PMID: 27919820 Review.
Cited by
-
Cancer- and endotoxin-induced cachexia require intact glucocorticoid signaling in skeletal muscle.FASEB J. 2013 Sep;27(9):3572-82. doi: 10.1096/fj.13-230375. Epub 2013 Jun 3. FASEB J. 2013. PMID: 23733748 Free PMC article.
-
Muscle cell and motor protein function in patients with a IIa myosin missense mutation (Glu-706 to Lys).Neuromuscul Disord. 2006 Nov;16(11):782-91. doi: 10.1016/j.nmd.2006.07.023. Epub 2006 Sep 26. Neuromuscul Disord. 2006. PMID: 17005402 Free PMC article.
-
Myoglobin plasma level related to muscle mass and fiber composition: a clinical marker of muscle wasting?J Mol Med (Berl). 2007 Aug;85(8):887-96. doi: 10.1007/s00109-007-0220-3. Epub 2007 Jun 30. J Mol Med (Berl). 2007. PMID: 17605115
-
Identification of squamous cell carcinoma associated proteins by proteomics and loss of beta tropomyosin expression in esophageal cancer.World J Gastroenterol. 2006 Nov 28;12(44):7104-12. doi: 10.3748/wjg.v12.i44.7104. World J Gastroenterol. 2006. PMID: 17131471 Free PMC article.
-
Emerging Mechanisms of Skeletal Muscle Homeostasis and Cachexia: The SUMO Perspective.Cells. 2023 Feb 17;12(4):644. doi: 10.3390/cells12040644. Cells. 2023. PMID: 36831310 Free PMC article. Review.
References
-
- Tisdale MJ. Biology of cachexia. J. Natl. Cancer Inst. 1997;89:1763–1773. - PubMed
-
- Argiles JM, Moore-Carrasco R, Fuster G, Busquets S, Lopez-Soriano FJ. Cancer cachexia: the molecular mechanisms. Int. J. Biochem. Cell Biol. 2003;35:405–409. - PubMed
-
- Tisdale MJ. Cachexia in cancer patients. Nat. Rev. Cancer. 2002;2:862–871. - PubMed
-
- Body JJ. The syndrome of anorexia-cachexia. Curr. Opin. Oncol. 1999;11:255–260. - PubMed
-
- Evans WK, et al. Limited impact of total parenteral nutrition on nutritional status during treatment for small cell lung cancer. Cancer Res. 1985;45:3347–3353. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
