

Nuclear factor of activated T cells balances angiogenesis activation and inhibition
- PMID: 15184502
- PMCID: PMC2211785
- DOI: 10.1084/jem.20040474
Nuclear factor of activated T cells balances angiogenesis activation and inhibition
Abstract
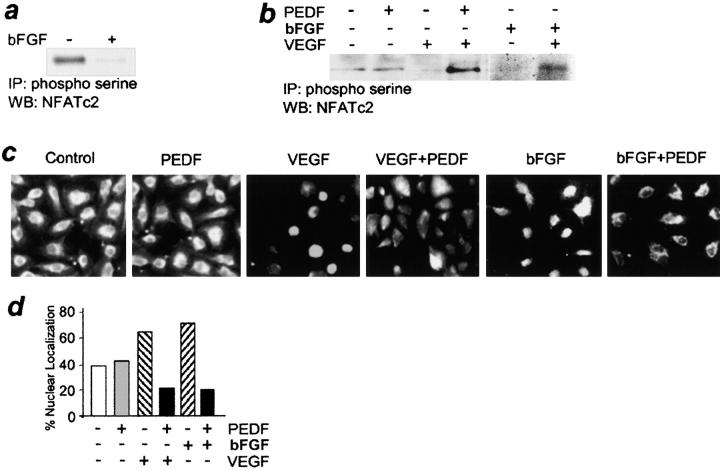
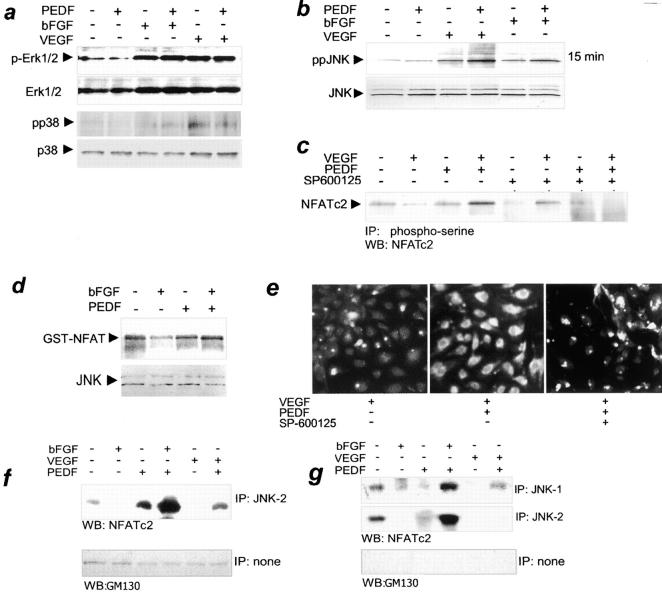
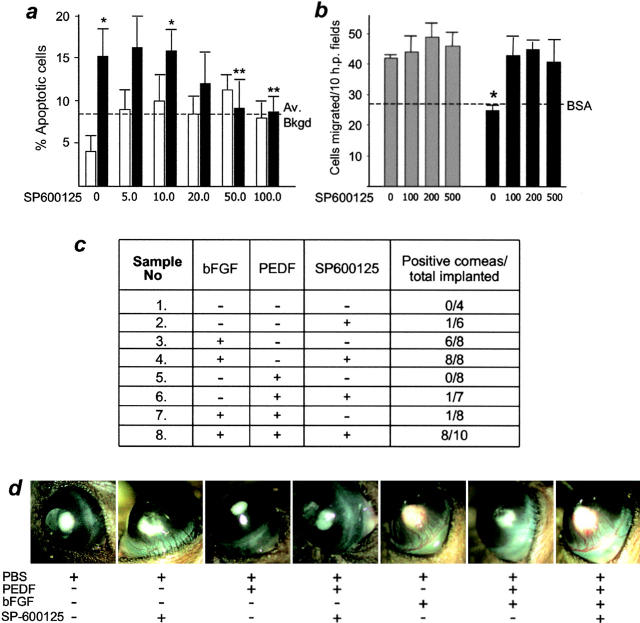
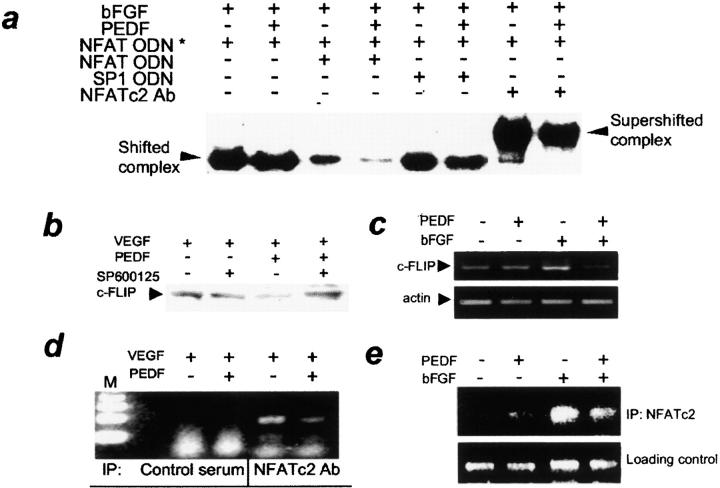
It has been demonstrated that vascular endothelial cell growth factor (VEGF) induction of angiogenesis requires activation of the nuclear factor of activated T cells (NFAT). We show that NFATc2 is also activated by basic fibroblast growth factor and blocked by the inhibitor of angiogenesis pigment epithelial-derived factor (PEDF). This suggests a pivotal role for this transcription factor as a convergence point between stimulatory and inhibitory signals in the regulation of angiogenesis. We identified c-Jun NH2-terminal kinases (JNKs) as essential upstream regulators of NFAT activity in angiogenesis. We distinguished JNK-2 as responsible for NFATc2 cytoplasmic retention by PEDF and JNK-1 and JNK-2 as mediators of PEDF-driven NFAT nuclear export. We identified a novel NFAT target, caspase-8 inhibitor cellular Fas-associated death domain-like interleukin 1beta-converting enzyme inhibitory protein (c-FLIP), whose expression was coregulated by VEGF and PEDF. Chromatin immunoprecipitation showed VEGF-dependent increase of NFATc2 binding to the c-FLIP promoter in vivo, which was attenuated by PEDF. We propose that one possible mechanism of concerted angiogenesis regulation by activators and inhibitors may be modulation of the endothelial cell apoptosis via c-FLIP controlled by NFAT and its upstream regulator JNK.
Figures
Similar articles
-
Two functional epitopes of pigment epithelial-derived factor block angiogenesis and induce differentiation in prostate cancer.Cancer Res. 2005 Jun 15;65(12):5144-52. doi: 10.1158/0008-5472.CAN-04-3744. Cancer Res. 2005. PMID: 15958558
-
Pigment epithelial-derived factor inhibits c-FLIP expression and assists ciglitazone-induced apoptosis in hepatocellular carcinoma.Anticancer Res. 2011 Apr;31(4):1173-80. Anticancer Res. 2011. PMID: 21508362
-
c-Jun N-terminal kinase (JNK) positively regulates NFATc2 transactivation through phosphorylation within the N-terminal regulatory domain.J Biol Chem. 2005 May 27;280(21):20867-78. doi: 10.1074/jbc.M501898200. Epub 2005 Mar 2. J Biol Chem. 2005. PMID: 15743762
-
Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs.Cardiovasc Res. 2004 Aug 15;63(3):467-75. doi: 10.1016/j.cardiores.2004.01.021. Cardiovasc Res. 2004. PMID: 15276472 Review.
-
Cell cycle and apoptosis regulation by NFAT transcription factors: new roles for an old player.Cell Death Dis. 2016 Apr 21;7(4):e2199. doi: 10.1038/cddis.2016.97. Cell Death Dis. 2016. PMID: 27100893 Free PMC article. Review.
Cited by
-
VEGF-A isoform-specific regulation of calcium ion flux, transcriptional activation and endothelial cell migration.Biol Open. 2015 Apr 24;4(6):731-42. doi: 10.1242/bio.201410884. Biol Open. 2015. PMID: 25910937 Free PMC article.
-
Pigment epithelium-derived factor blocks tumor extravasation by suppressing amoeboid morphology and mesenchymal proteolysis.Neoplasia. 2011 Jul;13(7):633-42. doi: 10.1593/neo.11446. Neoplasia. 2011. PMID: 21750657 Free PMC article.
-
Short pigment epithelial-derived factor-derived peptide inhibits angiogenesis and tumor growth.Clin Cancer Res. 2009 Mar 1;15(5):1655-63. doi: 10.1158/1078-0432.CCR-08-2113. Epub 2009 Feb 17. Clin Cancer Res. 2009. PMID: 19223494 Free PMC article.
-
The interleukin-1 receptor associated kinase 1 contributes to the regulation of NFAT.Mol Immunol. 2008 Sep;45(15):3902-8. doi: 10.1016/j.molimm.2008.06.023. Epub 2008 Aug 8. Mol Immunol. 2008. PMID: 18691762 Free PMC article.
-
Thrombospondin-1 and pigment epithelium-derived factor enhance responsiveness of KM12 colon tumor to metronomic cyclophosphamide but have disparate effects on tumor metastasis.Cancer Lett. 2013 Apr 28;330(2):241-9. doi: 10.1016/j.canlet.2012.11.055. Epub 2012 Dec 8. Cancer Lett. 2013. PMID: 23228633 Free PMC article.
References
-
- Bouck, N., V. Stellmach, and S.C. Hsu. 1996. How tumors become angiogenic. Adv. Cancer Res. 69:135–174. - PubMed
-
- Folkman, J. 2003. Angiogenesis and apoptosis. Semin. Cancer Biol. 13:159–167. - PubMed
-
- Bouck, N. 2002. PEDF: anti-angiogenic guardian of ocular function. Trends Mol. Med. 8:330–334. - PubMed
-
- Camphausen, K., and C. Menard. 2002. Angiogenesis inhibitors and radiotherapy of primary tumours. Expert Opin. Biol. Ther. 2:477–481. - PubMed
-
- Volpert, O.V., T. Zaichuk, W. Zhou, F. Reiher, T.A. Ferguson, P.M. Stuart, M. Amin, and N.P. Bouck. 2002. Inducer-stimulated Fas targets activated endothelium for destruction by anti-angiogenic thrombospondin-1 and pigment epithelium-derived factor. Nat. Med. 8:349–357. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
