

Cytoplasmic polyadenylation element binding protein-dependent protein synthesis is regulated by calcium/calmodulin-dependent protein kinase II
- PMID: 15175389
- PMCID: PMC6729187
- DOI: 10.1523/JNEUROSCI.0854-04.2004
Cytoplasmic polyadenylation element binding protein-dependent protein synthesis is regulated by calcium/calmodulin-dependent protein kinase II
Abstract
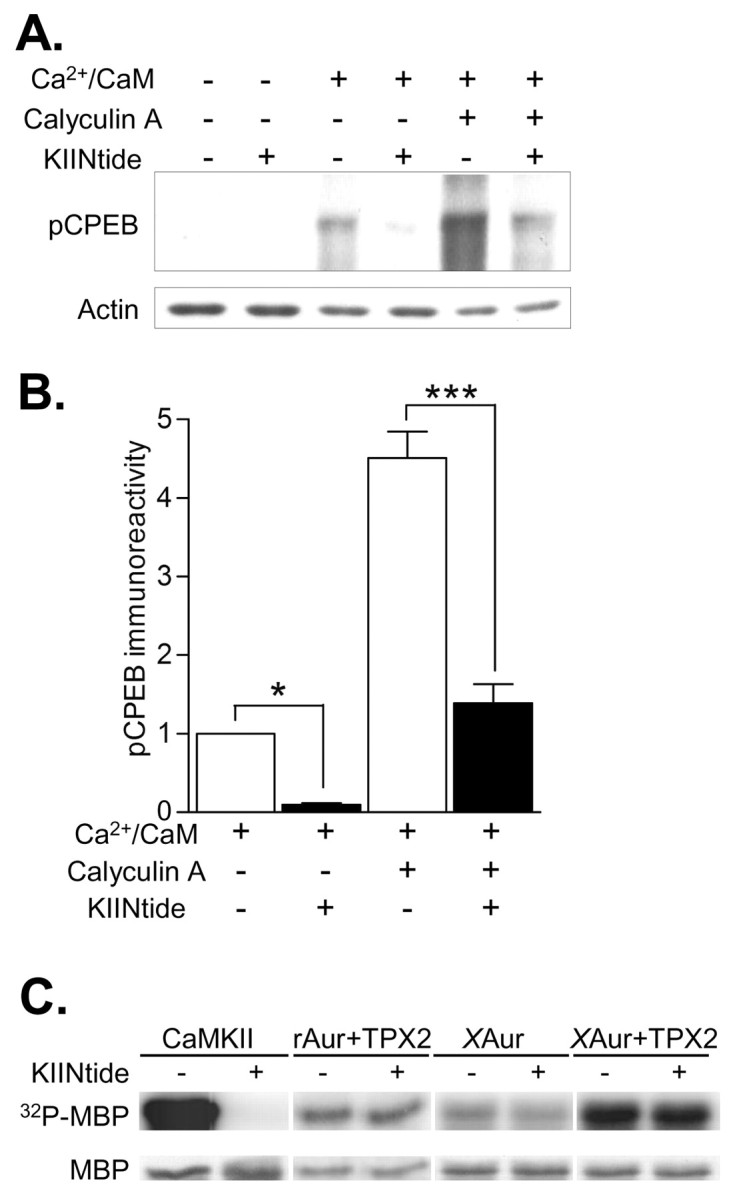
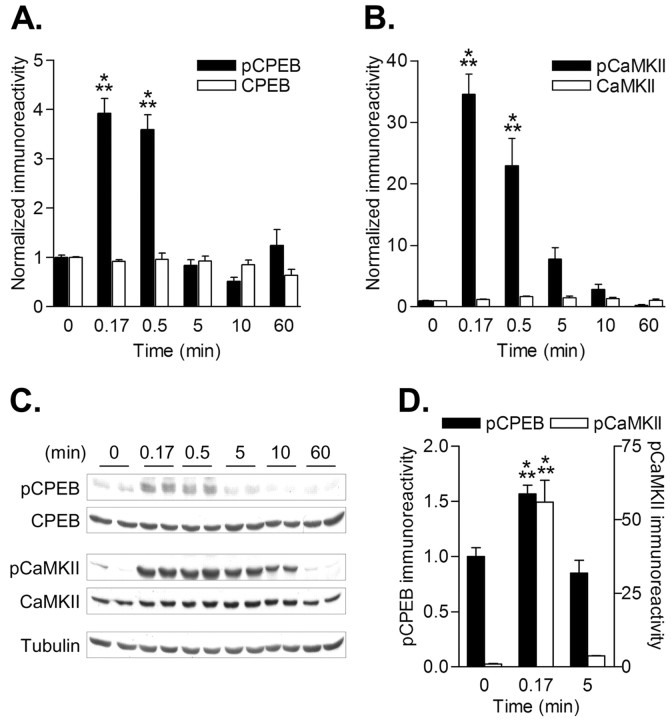
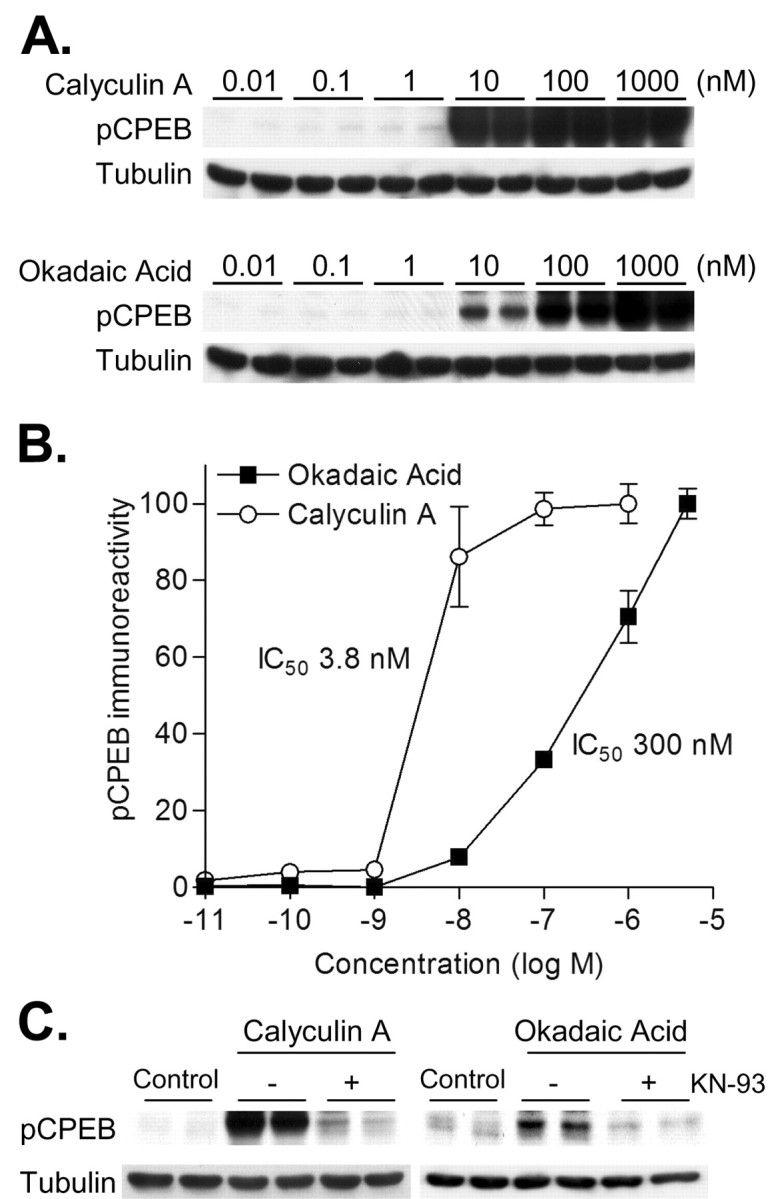
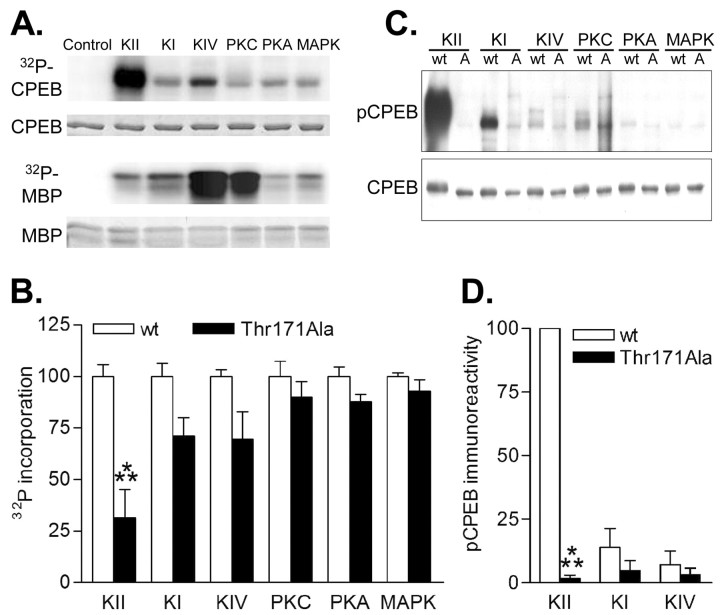
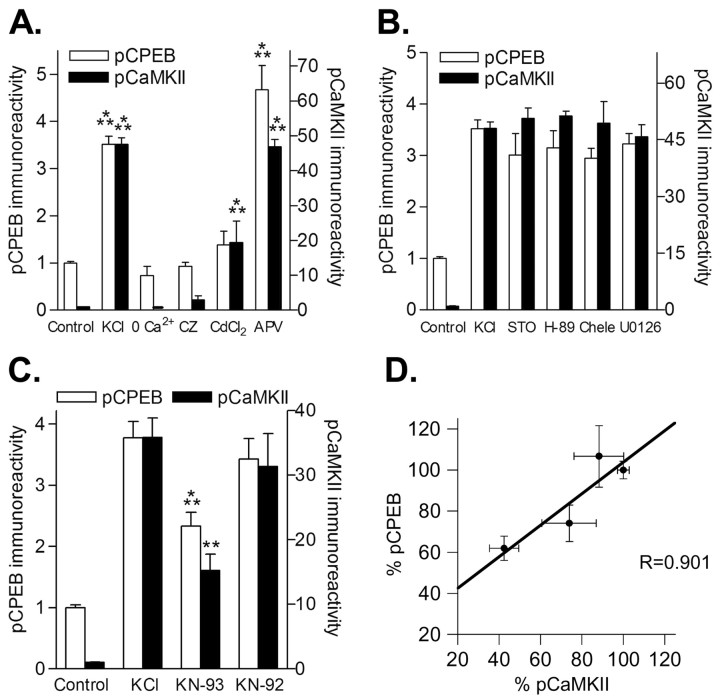
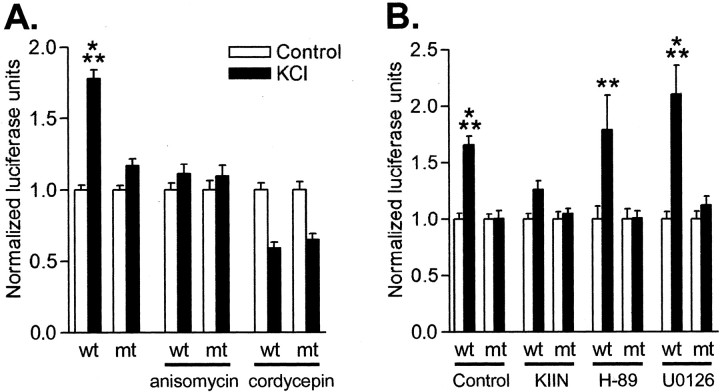
Phosphorylation of cytoplasmic polyadenylation element binding protein (CPEB) regulates protein synthesis in hippocampal dendrites. CPEB binds the 3' untranslated region (UTR) of cytoplasmic mRNAs and, when phosphorylated, initiates mRNA polyadenylation and translation. We report that, of the protein kinases activated in the hippocampus during synaptic plasticity, calcium/calmodulin-dependent protein kinase II (CaMKII) robustly phosphorylated the regulatory site (threonine 171) in CPEB in vitro. In postsynaptic density fractions or hippocampal neurons, CPEB phosphorylation increased when CaMKII was activated. These increases in CPEB phosphorylation were attenuated by a specific peptide inhibitor of CaMKII and by the general CaM-kinase inhibitor KN-93. Inhibitors of protein phosphatase 1 increased basal CPEB phosphorylation in neurons; this was also attenuated by a CaM-kinase inhibitor. To determine whether CaM-kinase activity regulates CPEB-dependent mRNA translation, hippocampal neurons were transfected with luciferase fused to a 3' UTR containing CPE-binding elements. Depolarization of neurons stimulated synthesis of luciferase; this was abrogated by inhibitors of protein synthesis, mRNA polyadenylation, and CaMKII. These results demonstrate that CPEB phosphorylation and translation are regulated by CaMKII activity and provide a possible mechanism for how dendritic protein synthesis in the hippocampus may be stimulated during synaptic plasticity.
Figures
Similar articles
-
Bidirectional regulation of cytoplasmic polyadenylation element-binding protein phosphorylation by Ca2+/calmodulin-dependent protein kinase II and protein phosphatase 1 during hippocampal long-term potentiation.J Neurosci. 2005 Jun 8;25(23):5604-10. doi: 10.1523/JNEUROSCI.5051-04.2005. J Neurosci. 2005. PMID: 15944388 Free PMC article.
-
CPEB-mediated cytoplasmic polyadenylation and the regulation of experience-dependent translation of alpha-CaMKII mRNA at synapses.Neuron. 1998 Nov;21(5):1129-39. doi: 10.1016/s0896-6273(00)80630-3. Neuron. 1998. PMID: 9856468
-
A molecular circuit composed of CPEB-1 and c-Jun controls growth hormone-mediated synaptic plasticity in the mouse hippocampus.J Neurosci. 2008 Aug 20;28(34):8502-9. doi: 10.1523/JNEUROSCI.1756-08.2008. J Neurosci. 2008. PMID: 18716208 Free PMC article.
-
Cataloguing and Selection of mRNAs Localized to Dendrites in Neurons and Regulated by RNA-Binding Proteins in RNA Granules.Biomolecules. 2020 Jan 22;10(2):167. doi: 10.3390/biom10020167. Biomolecules. 2020. PMID: 31978946 Free PMC article. Review.
-
Analysis of mRNA translation in cultured hippocampal neurons.Methods Enzymol. 2007;431:143-62. doi: 10.1016/S0076-6879(07)31008-2. Methods Enzymol. 2007. PMID: 17923234 Review.
Cited by
-
A model of the roles of essential kinases in the induction and expression of late long-term potentiation.Biophys J. 2006 Apr 15;90(8):2760-75. doi: 10.1529/biophysj.105.072470. Epub 2006 Jan 13. Biophys J. 2006. PMID: 16415049 Free PMC article.
-
RNA-binding proteins: a lesson in repression.J Neurosci. 2006 Jul 5;26(27):7135-8. doi: 10.1523/JNEUROSCI.1795-06.2006. J Neurosci. 2006. PMID: 16822967 Free PMC article. Review.
-
Bidirectional control of mRNA translation and synaptic plasticity by the cytoplasmic polyadenylation complex.Mol Cell. 2012 Jul 27;47(2):253-66. doi: 10.1016/j.molcel.2012.05.016. Epub 2012 Jun 21. Mol Cell. 2012. PMID: 22727665 Free PMC article.
-
Cytoplasmic Polyadenylation Element Binding Proteins CPEB1 and CPEB3 Regulate the Translation of FosB and Are Required for Maintaining Addiction-Like Behaviors Induced by Cocaine.Front Cell Neurosci. 2020 Jul 9;14:207. doi: 10.3389/fncel.2020.00207. eCollection 2020. Front Cell Neurosci. 2020. PMID: 32742260 Free PMC article.
-
Intracellular calcium and calmodulin link brain-derived neurotrophic factor to p70S6 kinase phosphorylation and dendritic protein synthesis.J Neurosci Res. 2010 May 15;88(7):1420-32. doi: 10.1002/jnr.22321. J Neurosci Res. 2010. PMID: 20029971 Free PMC article.
References
-
- Aakalu G, Smith WB, Nguyen N, Jiang C, Schuman EM (2001) Dynamic visualization of local protein synthesis in hippocampal neurons. Neuron 30: 489–502. - PubMed
-
- Blitzer RD, Connor JH, Brown GP, Wong T, Shenolikar S, Iyengar R, Landau EM (1998) Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science 280: 1940–1942. - PubMed
-
- Brewer GJ (1997) Isolation and culture of adult rat hippocampal neurons. J Neurosci Methods 71: 143–155. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous