

Cleavage between replicase proteins p28 and p65 of mouse hepatitis virus is not required for virus replication
- PMID: 15140993
- PMCID: PMC415798
- DOI: 10.1128/JVI.78.11.5957-5965.2004
Cleavage between replicase proteins p28 and p65 of mouse hepatitis virus is not required for virus replication
Abstract
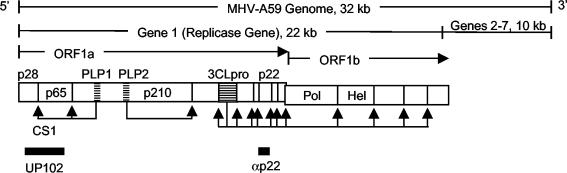
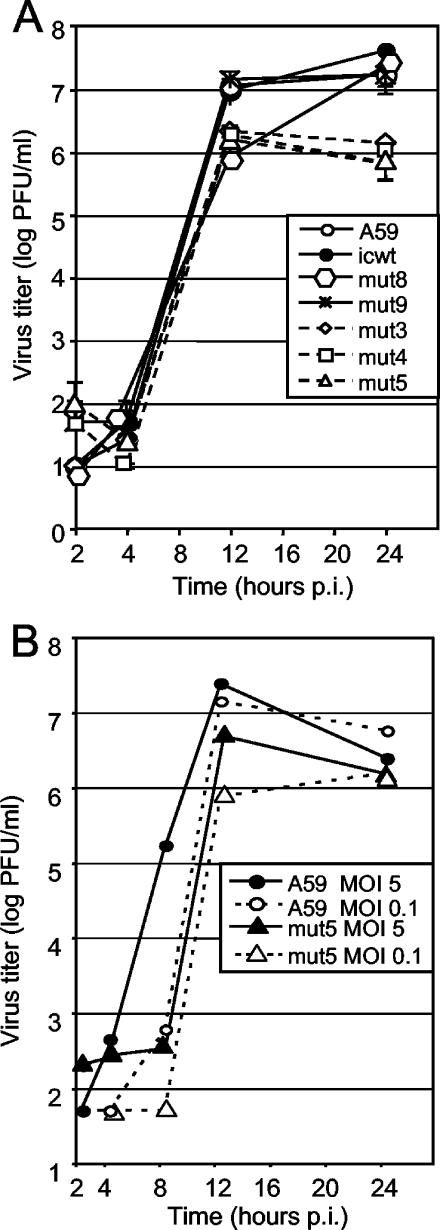
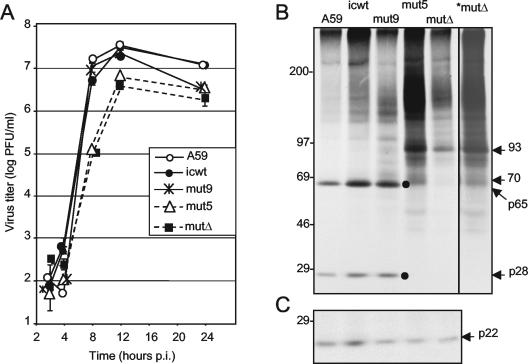
The p28 and p65 proteins of mouse hepatitis virus (MHV) are the most amino-terminal protein domains of the replicase polyprotein. Cleavage between p28 and p65 has been shown to occur in vitro at cleavage site 1 (CS1), (247)Gly downward arrow Val(248), in the polyprotein. Although critical residues for CS1 cleavage have been mapped in vitro, the requirements for cleavage have not been studied in infected cells. To define the determinants of CS1 cleavage and the role of processing at this site during MHV replication, mutations and deletions were engineered in the replicase polyprotein at CS1. Mutations predicted to allow cleavage at CS1 yielded viable virus that grew to wild-type MHV titers and showed normal expression and processing of p28 and p65. Mutant viruses containing predicted noncleaving mutations or a CS1 deletion were also viable but demonstrated delayed growth kinetics, reduced peak titers, decreased RNA synthesis, and small plaques compared to wild-type controls. No p28 or p65 was detected in cells infected with predicted noncleaving CS1 mutants or the CS1 deletion mutant; however, a new protein of 93 kDa was detected. All introduced mutations and the deletion were retained during repeated virus passages in culture, and no phenotypic reversion was observed. The results of this study demonstrate that cleavage between p28 and p65 at CS1 is not required for MHV replication. However, proteolytic separation of p28 from p65 is necessary for optimal RNA synthesis and virus growth, suggesting important roles for these proteins in the formation or function of viral replication complexes.
Figures




Similar articles
-
Identification of the polymerase polyprotein products p72 and p65 of the murine coronavirus MHV-JHM.Virus Res. 1996 Dec;45(2):101-9. doi: 10.1016/s0168-1702(96)01368-8. Virus Res. 1996. PMID: 8896245 Free PMC article.
-
Processing of open reading frame 1a replicase proteins nsp7 to nsp10 in murine hepatitis virus strain A59 replication.J Virol. 2007 Oct;81(19):10280-91. doi: 10.1128/JVI.00017-07. Epub 2007 Jul 18. J Virol. 2007. PMID: 17634238 Free PMC article.
-
Mouse hepatitis virus replicase proteins associate with two distinct populations of intracellular membranes.J Virol. 2000 Jun;74(12):5647-54. doi: 10.1128/jvi.74.12.5647-5654.2000. J Virol. 2000. PMID: 10823872 Free PMC article.
-
Proteolytic Processing of the Coronavirus Replicase Nonstructural Protein 14 Exonuclease Is Not Required for Virus Replication but Alters RNA Synthesis and Viral Fitness.J Virol. 2022 Aug 24;96(16):e0084122. doi: 10.1128/jvi.00841-22. Epub 2022 Aug 4. J Virol. 2022. PMID: 35924922 Free PMC article.
-
Mouse hepatitis virus replicase protein complexes are translocated to sites of M protein accumulation in the ERGIC at late times of infection.Virology. 2001 Jun 20;285(1):21-9. doi: 10.1006/viro.2001.0932. Virology. 2001. PMID: 11414802 Free PMC article.
Cited by
-
Murine hepatitis virus nonstructural protein 4 regulates virus-induced membrane modifications and replication complex function.J Virol. 2010 Jan;84(1):280-90. doi: 10.1128/JVI.01772-09. J Virol. 2010. PMID: 19846526 Free PMC article.
-
Atlas of coronavirus replicase structure.Virus Res. 2014 Dec 19;194:49-66. doi: 10.1016/j.virusres.2013.12.004. Epub 2013 Dec 16. Virus Res. 2014. PMID: 24355834 Free PMC article. Review.
-
The nsp2 replicase proteins of murine hepatitis virus and severe acute respiratory syndrome coronavirus are dispensable for viral replication.J Virol. 2005 Nov;79(21):13399-411. doi: 10.1128/JVI.79.21.13399-13411.2005. J Virol. 2005. PMID: 16227261 Free PMC article.
-
Murine coronaviruses encoding nsp2 at different genomic loci have altered replication, protein expression, and localization.J Virol. 2008 Dec;82(23):11964-9. doi: 10.1128/JVI.01126-07. Epub 2008 Sep 24. J Virol. 2008. PMID: 18815297 Free PMC article.
-
Nidovirales: evolving the largest RNA virus genome.Virus Res. 2006 Apr;117(1):17-37. doi: 10.1016/j.virusres.2006.01.017. Epub 2006 Feb 28. Virus Res. 2006. PMID: 16503362 Free PMC article. Review.
References
-
- Bonilla, P. J., S. A. Hughes, J. D. Pinon, and S. R. Weiss. 1995. Characterization of the leader papain-like proteinase of MHV-A59: identification of a new in vitro cleavage site. Virology 209:489-497. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
