

Neuronal pentraxin 1: a novel mediator of hypoxic-ischemic injury in neonatal brain
- PMID: 15115814
- PMCID: PMC6729280
- DOI: 10.1523/JNEUROSCI.0347-04.2004
Neuronal pentraxin 1: a novel mediator of hypoxic-ischemic injury in neonatal brain
Abstract
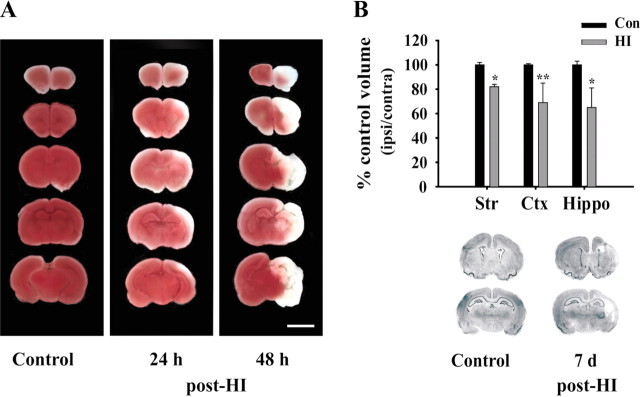
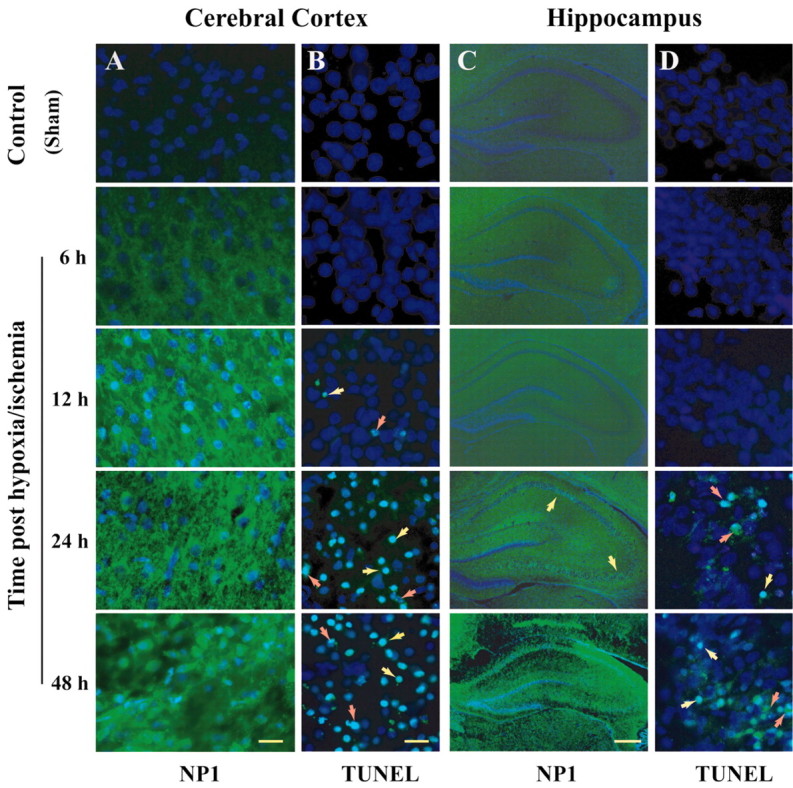
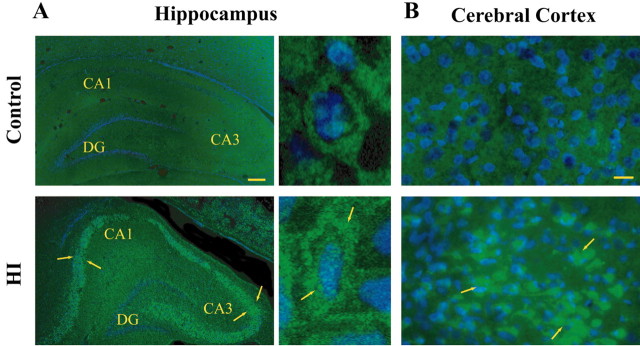
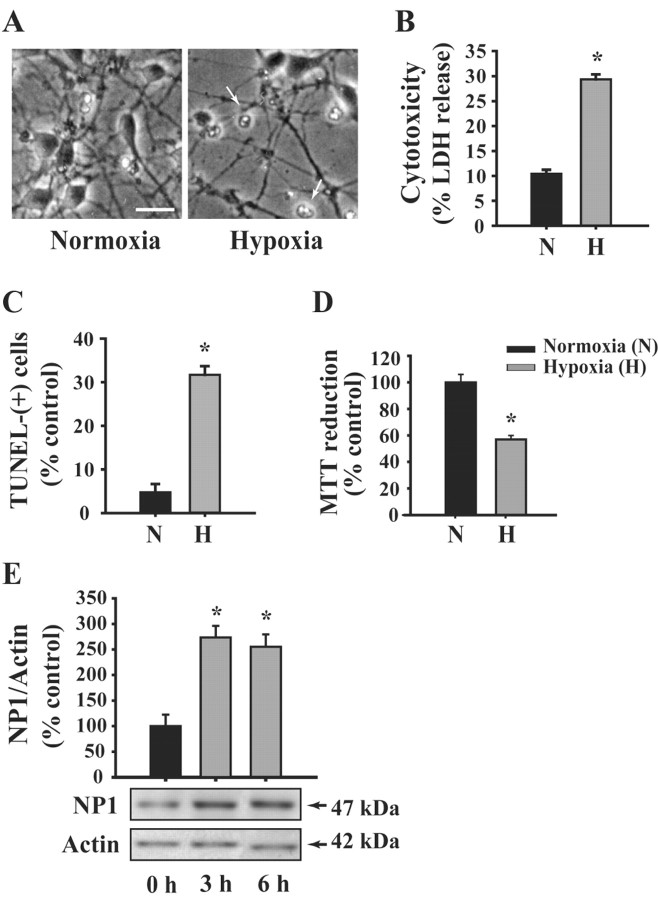
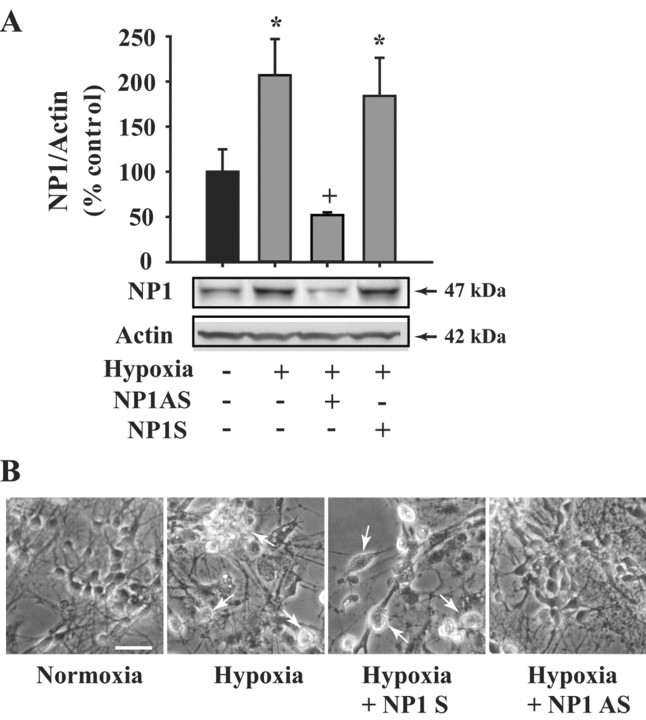
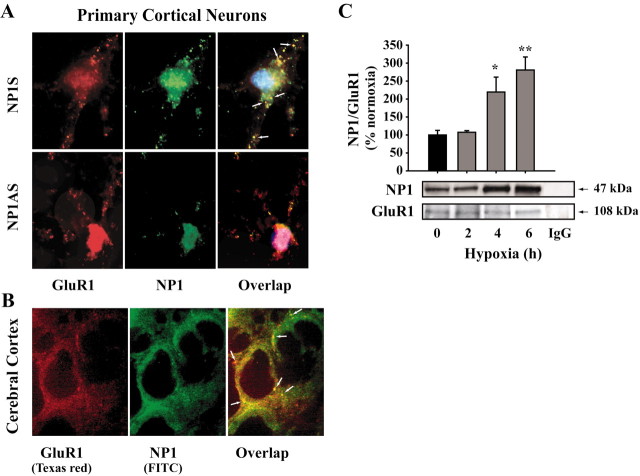
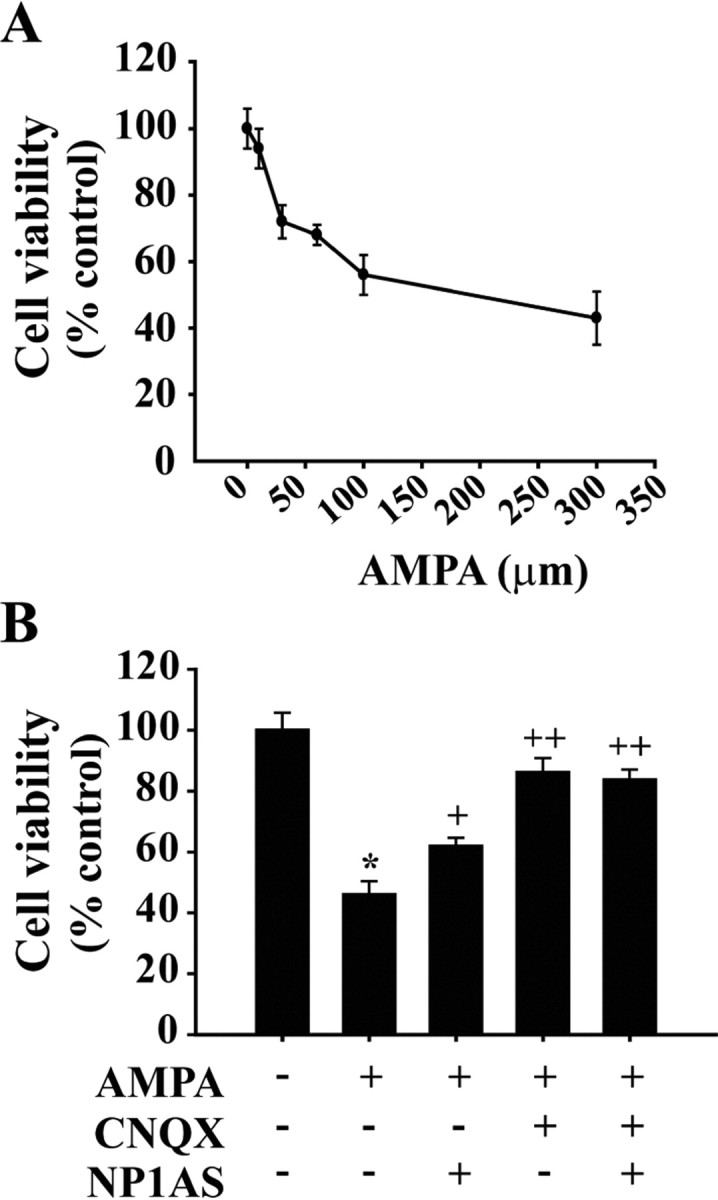
Neonatal hypoxic-ischemic brain injury is a major cause of neurological disability and mortality. Its therapy will likely require a greater understanding of the discrete neurotoxic molecular mechanism(s) triggered by hypoxia-ischemia (HI). Here, we investigated the role of neuronal pentraxin 1 (NP1), a member of a newly recognized subfamily of "long pentraxins," in the HI injury cascade. Neonatal brains developed marked infarcts in the ipsilateral cerebral hemisphere at 24 hr and showed significant loss of ipsilateral striatal, cortical, and hippocampal volumes at 7 d after HI compared with the contralateral hemisphere and sham controls. Immunofluorescence analyses revealed elevated neuronal expression of NP1 in the ipsilateral cerebral cortex from 6 hr to 7 d and in the hippocampal CA1 and CA3 regions from 24 hr to 7 d after HI. These same brain areas developed infarcts and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling-positive cells within 24-48 hr of HI. In primary cortical neurons, NP1 protein was induced >2.5-fold (p < 0.001) after their exposure to hypoxia that caused approximately 30-40% neuronal death. Transfecting cortical neurons with antisense oligodeoxyribonucleotides directed against NP1 mRNA (NP1AS) significantly inhibited (p < 0.01) hypoxia-induced NP1 protein induction and neuronal death (p < 0.001), demonstrating a specific requirement of NP1 in hypoxic neuronal injury. NP1 protein colocalized and coimmunoprecipitated with the fast excitatory AMPA glutamate receptor subunit (GluR1) in primary cortical neurons, and hypoxia induced a time-dependent increase in NP1-GluR1 interactions. NPIAS also protected against AMPA-induced neuronal death (p < 0.05), implicating a role for NP1 in the excitotoxic cascade. Our results show that NP1 induction mediates hypoxic-ischemic injury probably by interacting with and modulating GluR1 and potentially other excitatory glutamate receptors.
Figures

Similar articles
-
Critical role of extracellularly secreted neuronal pentraxin 1 in ischemic neuronal death.BMC Neurosci. 2014 Dec 20;15:133. doi: 10.1186/s12868-014-0133-3. BMC Neurosci. 2014. PMID: 25526743 Free PMC article.
-
Genetic deletion of neuronal pentraxin 1 expression prevents brain injury in a neonatal mouse model of cerebral hypoxia-ischemia.Neurobiol Dis. 2015 Mar;75:15-30. doi: 10.1016/j.nbd.2014.12.016. Epub 2014 Dec 29. Neurobiol Dis. 2015. PMID: 25554688 Free PMC article.
-
Hypoxic-ischemic injury in neonatal brain: involvement of a novel neuronal molecule in neuronal cell death and potential target for neuroprotection.Int J Dev Neurosci. 2008 Feb;26(1):93-101. doi: 10.1016/j.ijdevneu.2007.08.013. Epub 2007 Sep 7. Int J Dev Neurosci. 2008. PMID: 17936538 Free PMC article. Review.
-
Genetic deletion of NP1 prevents hypoxic-ischemic neuronal death via reducing AMPA receptor synaptic localization in hippocampal neurons.J Am Heart Assoc. 2013 Feb 22;2(1):e006098. doi: 10.1161/JAHA.112.006098. J Am Heart Assoc. 2013. PMID: 23525449 Free PMC article.
-
Molecular mediators of hypoxic-ischemic injury and implications for epilepsy in the developing brain.Epilepsy Behav. 2005 Sep;7(2):204-13. doi: 10.1016/j.yebeh.2005.05.015. Epilepsy Behav. 2005. PMID: 16054439 Review.
Cited by
-
Critical role of extracellularly secreted neuronal pentraxin 1 in ischemic neuronal death.BMC Neurosci. 2014 Dec 20;15:133. doi: 10.1186/s12868-014-0133-3. BMC Neurosci. 2014. PMID: 25526743 Free PMC article.
-
Sex differences in mitochondrial biogenesis determine neuronal death and survival in response to oxygen glucose deprivation and reoxygenation.BMC Neurosci. 2014 Jan 10;15:9. doi: 10.1186/1471-2202-15-9. BMC Neurosci. 2014. PMID: 24410996 Free PMC article.
-
Critical role of neuronal pentraxin 1 in mitochondria-mediated hypoxic-ischemic neuronal injury.Neurobiol Dis. 2013 Feb;50:59-68. doi: 10.1016/j.nbd.2012.10.003. Epub 2012 Oct 12. Neurobiol Dis. 2013. PMID: 23069675 Free PMC article.
-
Isobaric tags for relative and absolute quantitation-based quantitative proteomic analysis of X-linked inhibitor of apoptosis and H2AX in etoposide-induced renal cell carcinoma apoptosis.Chin Med J (Engl). 2019 Dec 20;132(24):2941-2949. doi: 10.1097/CM9.0000000000000553. Chin Med J (Engl). 2019. PMID: 31855962 Free PMC article.
-
Neuronal Pentraxin 1 Promotes Hypoxic-Ischemic Neuronal Injury by Impairing Mitochondrial Biogenesis via Interactions With Active Bax[6A7] and Mitochondrial Hexokinase II.ASN Neuro. 2021 Jan-Dec;13:17590914211012888. doi: 10.1177/17590914211012888. ASN Neuro. 2021. PMID: 34098747 Free PMC article.
References
-
- Barks JD, Silverstein FS (1992) Excitatory amino acids contribute to the pathogenesis of perinatal hypoxic-ischemic brain injury. Brain Pathol 2: 235-243. - PubMed
-
- Baude A, Nusser Z, Molnar E, McIlhinney RA, Somogyi P (1995) High-resolution immunogold localization of AMPA type glutamate receptor subunits at synaptic and non-synaptic sites in rat hippocampus. Neuroscience 69: 1031-1055. - PubMed
-
- Bittigau P, Sifringer M, Pohl D, Stadthaus D, Ishimaru M, Shimizu H, Ikeda M, Lang D, Speer A, Olney JW, Ikonomidou C (1999) Apoptotic neurodegeneration following trauma is markedly enhanced in the immature brain. Ann Neurol 45: 724-735. - PubMed
-
- Bottazzi B, Vouret-Craviari V, Bastone A, De Gioia L, Matteucci C, Peri G, Spreafico F, Pausa M, D'Ettorre C, Gianazza E, Tagliabue A, Salmona M, Tedesco F, Introna M, Mantovani A (1997) Multimer formation and ligand recognition by the long pentraxin PTX3. Similarities and differences with the short pentraxins C-reactive protein and serum amyloid P component. J Biol Chem 272: 32817-32823. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous