

Phylogenomics and bioinformatics of SARS-CoV
- PMID: 15058277
- PMCID: PMC7119090
- DOI: 10.1016/j.tim.2004.01.005
Phylogenomics and bioinformatics of SARS-CoV
Abstract
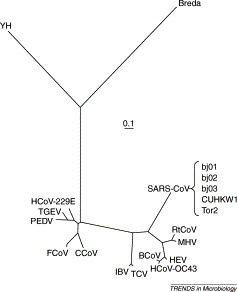
Tracing the history of molecular changes in coronaviruses using phylogenetic methods can provide powerful insights into the patterns of modification to sequences that underlie alteration to selective pressure and molecular function in the SARS-CoV (severe acute respiratory syndrome coronavirus) genome. The topology and branch lengths of the phylogenetic relationships among the family Coronaviridae, including SARS-CoV, have been estimated using the replicase polyprotein. The spike protein fragments S1 (involved in receptor-binding) and S2 (involved in membrane fusion) have been found to have different mutation rates. Fragment S1 can be further divided into two regions (S1A, which comprises approximately the first 400 nucleotides, and S1B, comprising the next 280) that also show different rates of mutation. The phylogeny presented on the basis of S1B shows that SARS-CoV is closely related to MHV (murine hepatitis virus), which is known to bind the murine receptor CEACAM1. The predicted structure, accessibility and mutation rate of the S1B region is also presented. Because anti-SARS drugs based on S2 heptads have short half-lives and are difficult to manufacture, our findings suggest that the S1B region might be of interest for anti-SARS drug discovery.
Figures

Similar articles
-
Phylogeny of the SARS coronavirus.Science. 2003 Nov 28;302(5650):1504-5. doi: 10.1126/science.302.5650.1504b. Science. 2003. PMID: 14645828 No abstract available.
-
Characterization of a novel coronavirus associated with severe acute respiratory syndrome.Science. 2003 May 30;300(5624):1394-9. doi: 10.1126/science.1085952. Epub 2003 May 1. Science. 2003. PMID: 12730500
-
Coronavirus genomic-sequence variations and the epidemiology of the severe acute respiratory syndrome.N Engl J Med. 2003 Jul 10;349(2):187-8. doi: 10.1056/NEJM200307103490216. N Engl J Med. 2003. PMID: 12853594 No abstract available.
-
Insights from the association of SARS-CoV S-protein with its receptor, ACE2.Adv Exp Med Biol. 2006;581:209-18. doi: 10.1007/978-0-387-33012-9_36. Adv Exp Med Biol. 2006. PMID: 17037532 Free PMC article. Review. No abstract available.
-
The spike protein of SARS-CoV--a target for vaccine and therapeutic development.Nat Rev Microbiol. 2009 Mar;7(3):226-36. doi: 10.1038/nrmicro2090. Epub 2009 Feb 9. Nat Rev Microbiol. 2009. PMID: 19198616 Free PMC article. Review.
Cited by
-
Electromagnetically-driven integrated microfluidic platform using reverse transcription loop-mediated isothermal amplification for detection of severe acute respiratory syndrome coronavirus 2.Anal Chim Acta. 2022 Aug 1;1219:340036. doi: 10.1016/j.aca.2022.340036. Epub 2022 Jun 6. Anal Chim Acta. 2022. PMID: 35715135 Free PMC article.
-
Evolution, Ecology, and Zoonotic Transmission of Betacoronaviruses: A Review.Front Vet Sci. 2021 May 20;8:644414. doi: 10.3389/fvets.2021.644414. eCollection 2021. Front Vet Sci. 2021. PMID: 34095271 Free PMC article. Review.
-
Bioinformatic prediction of immunodominant regions in spike protein for early diagnosis of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).PeerJ. 2021 Apr 8;9:e11232. doi: 10.7717/peerj.11232. eCollection 2021. PeerJ. 2021. PMID: 33889450 Free PMC article.
-
A human coronavirus evolves antigenically to escape antibody immunity.PLoS Pathog. 2021 Apr 8;17(4):e1009453. doi: 10.1371/journal.ppat.1009453. eCollection 2021 Apr. PLoS Pathog. 2021. PMID: 33831132 Free PMC article.
-
Pathogenetic profiling of COVID-19 and SARS-like viruses.Brief Bioinform. 2021 Mar 22;22(2):1175-1196. doi: 10.1093/bib/bbaa173. Brief Bioinform. 2021. PMID: 32778874 Free PMC article.
References
-
- Whelan S. Molecular phylogenetics: state-of-the-art methods for looking into the past. Trends Genet. 2001;17:262–272. - PubMed
-
- Marra M.A. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. - PubMed
-
- Rota P.A. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. - PubMed
-
- Russell R.B. Recognition of analogous and homologous protein folds: analysis of sequence and structure conservation. J. Mol. Biol. 1997;269:423–439. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
