

Molecular epidemiology of the novel coronavirus that causes severe acute respiratory syndrome
- PMID: 14726162
- PMCID: PMC7112497
- DOI: 10.1016/s0140-6736(03)15259-2
Molecular epidemiology of the novel coronavirus that causes severe acute respiratory syndrome
Abstract
Background: Severe acute respiratory syndrome (SARS) is a newly emerged disease caused by a novel coronavirus (SARS-CoV), which spread globally in early 2003, affecting over 30 countries. We have used molecular epidemiology to define the patterns of spread of the virus in Hong Kong and beyond.
Methods: The case definition of SARS was based on that recommended by WHO. We genetically sequenced the gene for the S1 unit of the viral spike protein of viruses from patients with SARS in Hong Kong (138) and Guangdong (three) in February to April, 2003. We undertook phylogenetic comparisons with 27 other sequences available from public databases (Genbank).
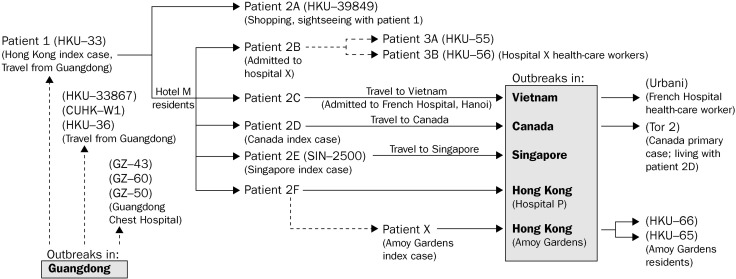
Findings: Most of the Hong Kong viruses (139/142), including those from a large outbreak in an apartment block, clustered closely together with the isolate from a single index case (HKU-33) who came from Guangdong to Hong Kong in late February. Three other isolates were genetically distinct from HKU-33 in Hong Kong during February, but none of these contributed substantially to the subsequent local outbreak. Viruses identified in Guangdong and Beijing were genetically more diverse.
Interpretation: The molecular epidemiological evidence suggests that most SARS-CoV from the outbreak in Hong Kong, as well as the viruses from Canada, Vietnam, and Singapore, are genetically closely linked. Three viruses found in Hong Kong in February were phylogenetically distinct from the major cluster, which suggests that several introductions of the virus had occurred, but that only one was associated with the subsequent outbreak in Hong Kong, which in turn spread globally.
Figures


Similar articles
-
Genomic characterisation of the severe acute respiratory syndrome coronavirus of Amoy Gardens outbreak in Hong Kong.Lancet. 2003 Nov 29;362(9398):1807-8. doi: 10.1016/s0140-6736(03)14901-x. Lancet. 2003. PMID: 14654320 Free PMC article.
-
Epidemiology of severe acute respiratory syndrome (SARS): adults and children.Paediatr Respir Rev. 2004 Dec;5(4):270-4. doi: 10.1016/j.prrv.2004.07.011. Paediatr Respir Rev. 2004. PMID: 15531250 Free PMC article. Review.
-
[Severe acute respiratory syndrome].Med Clin (Barc). 2003 May 3;120(16):626-9. doi: 10.1016/s0025-7753(03)73791-9. Med Clin (Barc). 2003. PMID: 12732129 Free PMC article. Spanish. No abstract available.
-
Severe acute respiratory syndrome (SARS): over 100 days into the outbreak.Wkly Epidemiol Rec. 2003 Jun 27;78(26):217-20. Wkly Epidemiol Rec. 2003. PMID: 15571170 English, French. No abstract available.
-
Severe acute respiratory syndrome epidemic in Taiwan, 2003.J Microbiol Immunol Infect. 2005 Apr;38(2):82-8. J Microbiol Immunol Infect. 2005. PMID: 15843851 Review.
Cited by
-
Forecasting mortality and DALYs from air pollution in SAARC nations.Sci Rep. 2024 Oct 29;14(1):25898. doi: 10.1038/s41598-024-76760-9. Sci Rep. 2024. PMID: 39468227 Free PMC article.
-
Modeling health outcomes of air pollution in the Middle East by using support vector machines and neural networks.Sci Rep. 2024 Sep 14;14(1):21517. doi: 10.1038/s41598-024-71694-8. Sci Rep. 2024. PMID: 39277668 Free PMC article.
-
Coronavirus and co-infections: A Saudi Arabian perspective.Saudi J Biol Sci. 2023 Sep;30(9):103739. doi: 10.1016/j.sjbs.2023.103739. Epub 2023 Jul 13. Saudi J Biol Sci. 2023. PMID: 37520787 Free PMC article. Review.
-
Deciphering molecular mechanisms of SARS-CoV-2 pathogenesis and drug repurposing through GRN motifs: a comprehensive systems biology study.3 Biotech. 2023 Apr;13(4):117. doi: 10.1007/s13205-023-03518-x. Epub 2023 Mar 13. 3 Biotech. 2023. PMID: 37070032 Free PMC article.
-
Diagnosis of SARS-CoV-2 during the Pandemic by Multiplex RT-rPCR hCoV Test: Future Perspectives.Pathogens. 2022 Nov 18;11(11):1378. doi: 10.3390/pathogens11111378. Pathogens. 2022. PMID: 36422629 Free PMC article.
References
-
- Ksiazek TG, Erdman D, Goldsmith CS. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med. 2003;348:1953–1956. - PubMed
-
- Centers for Disease Control and Prevention Update: outbreak of severe acute respiratory syndrome—worldwide. MMWR Morb Mortal Weekly Rep. 2003;52:241–245. - PubMed
-
- WHO Cumulative number of reported probable cases of severe acute respiratory syndrome (SARS) http://www.who.int/csr/sars/country/en (accessed Aug 25, 2003)
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
