

Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia
- PMID: 14715958
- PMCID: PMC6729582
- DOI: 10.1523/JNEUROSCI.4485-03.2004
Neuroprotective function of the PGE2 EP2 receptor in cerebral ischemia
Abstract
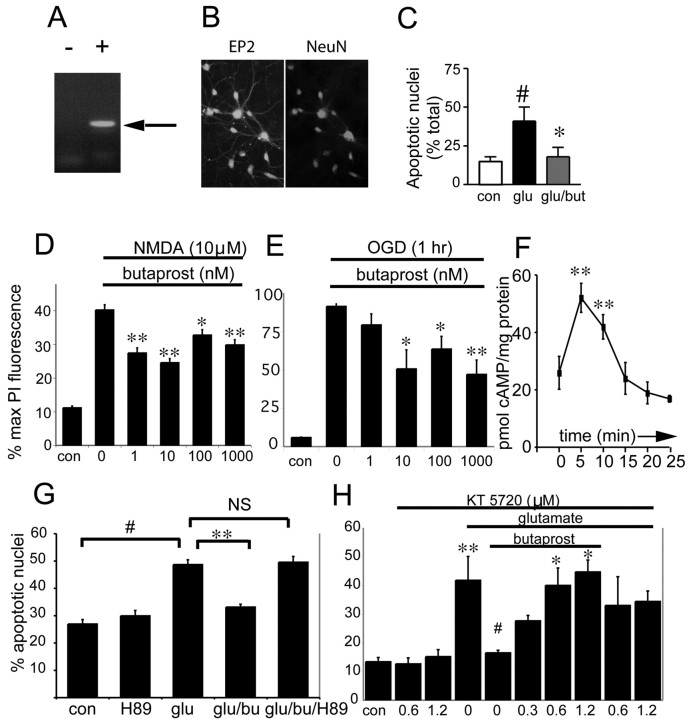
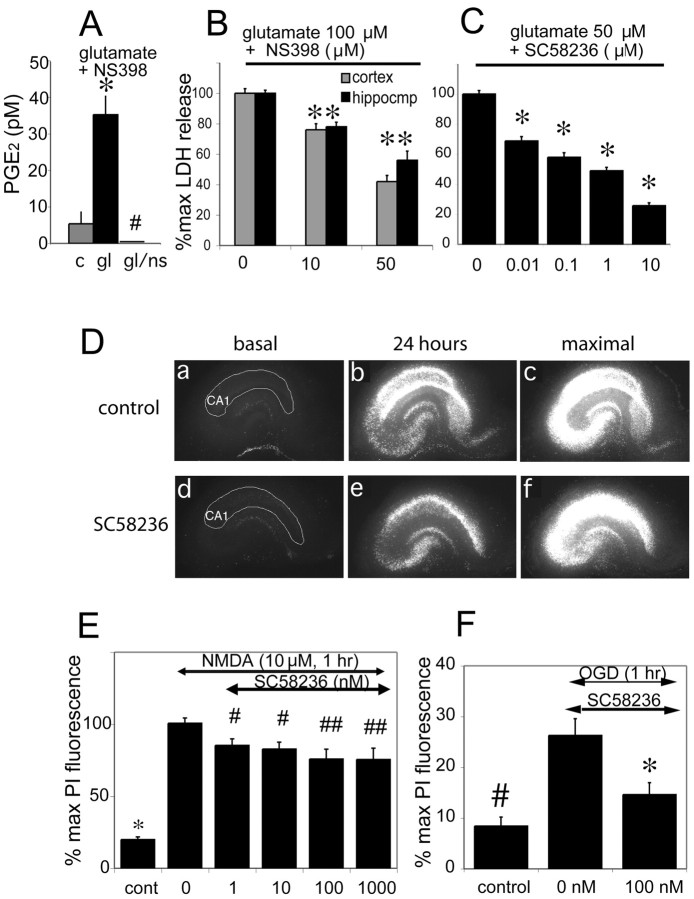
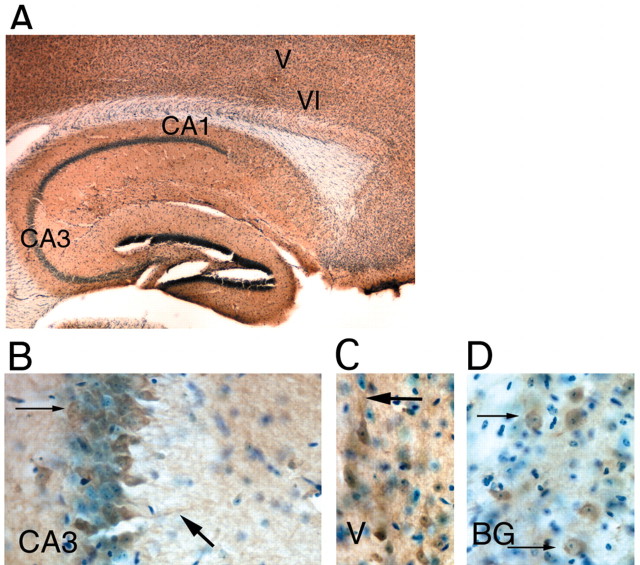
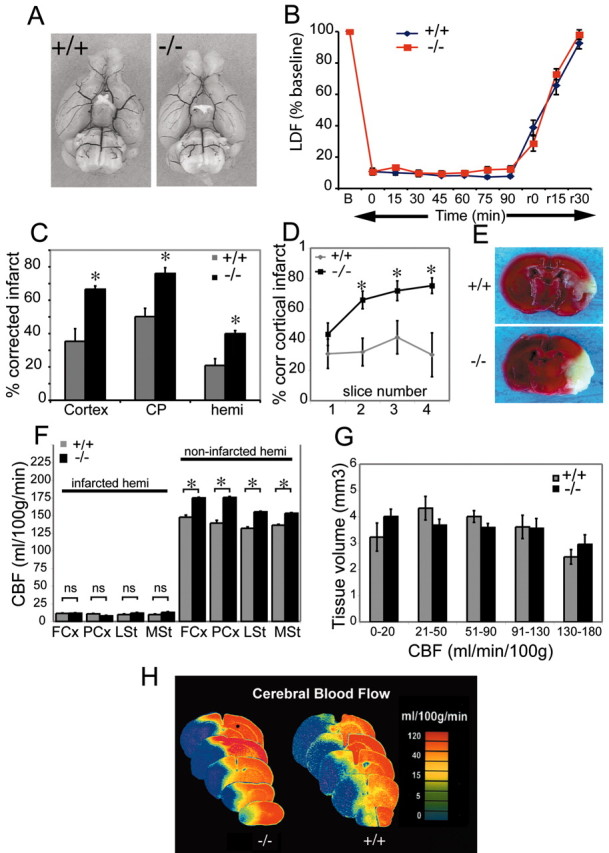
The cyclooxygenases COX-1 and COX-2 catalyze the first committed step of prostaglandin synthesis from arachidonic acid. Previous studies in rodent stroke models have shown that the inducible COX-2 isoform promotes neuronal injury, and the administration of COX-2 inhibitors reduces infarct volume. We investigated the function of PGE2, a principal prostaglandin product of COX-2 enzymatic activity, in neuronal survival in cerebral ischemia. PGE2 exerts its downstream effects by signaling through a class of four distinct G-protein-coupled EP receptors (for E-prostanoid: EP1, EP2, EP3, and EP4) that have divergent effects on cAMP and phosphoinositol turnover and different anatomical distributions in brain. The EP2 receptor subtype is abundantly expressed in cerebral cortex, striatum, and hippocampus, and is positively coupled to cAMP production. In vitro studies of dispersed neurons and organotypic hippocampal cultures demonstrated that activation of the EP2 receptor was neuroprotective in paradigms of NMDA toxicity and oxygen glucose deprivation. Pharmacologic blockade of EP2 signaling by inhibition of protein kinase A activation reversed this protective effect, suggesting that EP2-mediated neuroprotection is dependent on cAMP signaling. In the middle cerebral artery occlusion-reperfusion model of transient forebrain ischemia, genetic deletion of the EP2 receptor significantly increased cerebral infarction in cerebral cortex and subcortical structures. These studies indicate that activation of the PGE2 EP2 receptor can protect against excitotoxic and anoxic injury in a cAMP-dependent manner. Taken together, these data suggest a novel mechanism of neuroprotection mediated by a dominant PGE2 receptor subtype in brain that may provide a target for therapeutic intervention.
Figures

Similar articles
-
Stimulation of PGE receptors EP2 and EP4 protects cultured neurons against oxidative stress and cell death following beta-amyloid exposure.Eur J Neurosci. 2005 Nov;22(9):2199-206. doi: 10.1111/j.1460-9568.2005.04427.x. Eur J Neurosci. 2005. PMID: 16262658
-
Neuroprotection by the PGE2 EP2 receptor in permanent focal cerebral ischemia.Ann Neurol. 2005 May;57(5):758-61. doi: 10.1002/ana.20461. Ann Neurol. 2005. PMID: 15852374
-
PGE2 receptors rescue motor neurons in a model of amyotrophic lateral sclerosis.Ann Neurol. 2004 Aug;56(2):240-8. doi: 10.1002/ana.20179. Ann Neurol. 2004. PMID: 15293276
-
[Cooperation of two subtypes of PGE2 receptor, Gi coupled EP3 and Gs coupled EP2 or EP4 subtype].Yakugaku Zasshi. 2003 Oct;123(10):837-43. doi: 10.1248/yakushi.123.837. Yakugaku Zasshi. 2003. PMID: 14577329 Review. Japanese.
-
Regulation of prostanoid synthesis in microglial cells and effects of prostaglandin E2 on microglial functions.Biochimie. 1998 Nov;80(11):899-904. doi: 10.1016/s0300-9084(00)88886-0. Biochimie. 1998. PMID: 9893949 Review.
Cited by
-
Ultrasound stimulates formation and release of vasoactive compounds in brain endothelial cells.Am J Physiol Heart Circ Physiol. 2015 Aug 15;309(4):H583-91. doi: 10.1152/ajpheart.00690.2014. Epub 2015 Jun 19. Am J Physiol Heart Circ Physiol. 2015. PMID: 26092990 Free PMC article.
-
Skeletal interoception in bone homeostasis and pain.Cell Metab. 2022 Dec 6;34(12):1914-1931. doi: 10.1016/j.cmet.2022.09.025. Epub 2022 Oct 17. Cell Metab. 2022. PMID: 36257317 Free PMC article. Review.
-
New tricks by an old dogma: mechanisms of the Organizational/Activational Hypothesis of steroid-mediated sexual differentiation of brain and behavior.Horm Behav. 2009 May;55(5):655-65. doi: 10.1016/j.yhbeh.2009.02.012. Horm Behav. 2009. PMID: 19682425 Free PMC article. Review.
-
Cognitive outcome and cyclo-oxygenase-2 gene (-765 G/C) variation in the preterm infant.Arch Dis Child Fetal Neonatal Ed. 2007 Mar;92(2):F108-12. doi: 10.1136/adc.2006.099499. Epub 2006 Aug 11. Arch Dis Child Fetal Neonatal Ed. 2007. PMID: 16905570 Free PMC article.
-
Neuroprotection by selective allosteric potentiators of the EP2 prostaglandin receptor.Proc Natl Acad Sci U S A. 2010 Feb 2;107(5):2307-12. doi: 10.1073/pnas.0909310107. Epub 2010 Jan 14. Proc Natl Acad Sci U S A. 2010. PMID: 20080612 Free PMC article.
References
-
- Abe H, Takeshita T, Nagata K, Arita T, Endo Y, Fujita T, Takayama H, Kubo M, Sugamura K (1999) Molecular cloning, chromosome mapping and characterization of the mouse CRTH2 gene, a putative member of the leukocyte chemoattractant receptor family. Gene 227: 71-77. - PubMed
-
- Adams J, Collaco-Moraes Y, de Belleroche J (1996) Cyclooxygenase-2 induction in cerebral cortex: an intracellular response to synaptic excitation. J Neurochem 66: 6-13. - PubMed
-
- Akaike A, Kaneko S, Tamura Y, Nakata N, Shiomi H, Ushikubi F, Narumiya S (1994) Prostaglandin E2 protects cultured cortical neurons against N-methyl-d-aspartate receptor-mediated glutamate cytotoxcity. Brain Res 663: 237-244. - PubMed
-
- Alkayed NJ, Harakumi I, Kimes AS, London ED, Traystman RJ, Hurn PD (1998) Gender-linked injury in experimental stroke. Stroke 29: 159-165. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials