

doi: 10.1073/pnas.0307203101.
Epub 2003 Dec 29.
Somatic activation of oncogenic Kras in hematopoietic cells initiates a rapidly fatal myeloproliferative disorder
Affiliations
- PMID: 14699048
- PMCID: PMC327193
- DOI: 10.1073/pnas.0307203101
Item in Clipboard
Somatic activation of oncogenic Kras in hematopoietic cells initiates a rapidly fatal myeloproliferative disorder
Proc Natl Acad Sci U S A.
.
Abstract
RAS mutations are common in myeloid malignancies; however, it is not known whether oncogenic RAS can initiate leukemia. We show that expressing mutant K-Ras(G12D) protein from the endogenous murine locus rapidly induces a fatal myeloproliferative disorder with 100% penetrance characterized by tissue infiltration, hypersensitivity to growth factors, and hyperproliferation. Hematopoietic cells from diseased mice demonstrated increased levels of Ras-GTP, but effector kinases were not constitutively phosphorylated and responded normally to growth factors. Oncogenic RAS is sufficient to initiate myeloid leukemogenesis in mice, and this provides an in vivo system for biologic and preclinical studies.
Figures
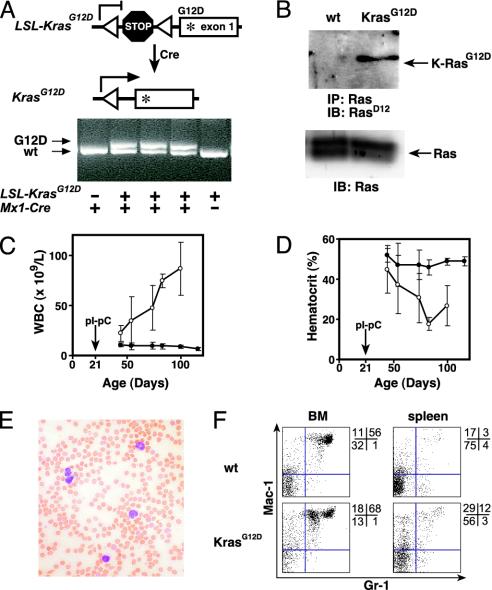
K-RasG12D expression induces MPD in Mx1-Cre KrasG12D mice. (A) Excision of the stop cassette by Cre recombinase shown in schematic (Upper) and by PCR (Lower) using primers flanking the loxP sites (triangles). Under these conditions, the wild type (KrasWT) and mutant (KrasG12D) give the indicated products, and the unrearranged allele does not amplify. PCR was performed on DNA purified from peripheral blood from mice with the indicated genotypes 3 weeks after injection with pIpC. (B) Expression of K-RasG12D protein in Mx1-Cre KrasG12D splenocytes but not those from a littermate control after treatment with pIpC. Immunoprecipitation of total Ras was followed by immunoblotting with an antibody specific for the K-RasG12D protein (Upper). Immunoblot of total cell lysates (Lower) demonstrates equal loading. (C and D) Progressive leukocytosis (C) and anemia (D) in a cohort of Mx1-Cre KrasG12D mice (n = 11; open circles) and littermate controls (n = 19; filled circles) that had serial blood counts performed after pIpC injection at weaning (±SEM). (E) Blood smear showing leukocytosis with mature myeloid cells in mice with MPD. (F) Flow cytometry of bone marrow and spleen cells from wild-type and Mx1-Cre KrasG12D mice, 7 weeks after pIpC injection. Proportions of cells within each quadrant are shown to the right of each plot.
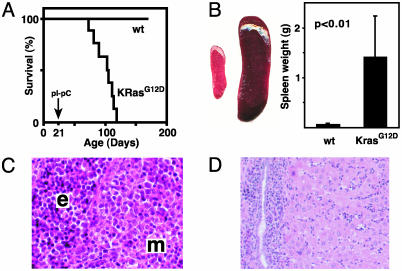
Survival and pathologic features of MPD in Mx1-Cre KrasG12D mice. (A) Kaplan–Meier survival curve in a cohort of Mx1-Cre KrasG12D (KrasG12D; n = 11) and littermate control wild-type (wt; n = 19) mice. Mx1-Cre KrasG12D mice have a median lifespan of 105 days (84 days after pIpC treatment), and all of the littermates were alive after 1 year of observation. The 100% penetrance of MPD was confirmed in 20 additional Mx1-Cre KrasG12D mice. (B) Massive splenomegaly, with a typical example of wild-type (Left) and Mx1-Cre KrasG12D (Right) spleens, and a graphical comparison of means and standard deviations (n = 19 Mx1-Cre KrasG12D vs. 17 control specimens) of spleen weights. (C and D) Hematoxylin and eosin-stained sections of Mx1-Cre KrasG12D spleen (C) and liver (D), showing extensive myeloid infiltration (m) and splenic erythroid hyperplasia (e).
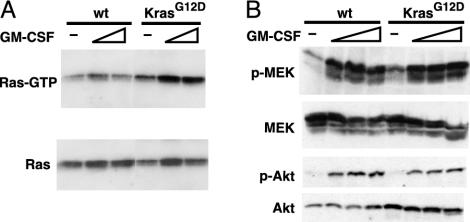
Ras activation and signaling in Mx1-Cre KrasG12D bone marrow. (A) Bone marrow cells from wild-type or Mx1-Cre KrasG12D mice were starved for 4 h in Iscove's modified Dulbecco's medium with 0.1% FCS, then stimulated for 3minwith1or3ng/ml GM-CSF. Ras-GTP was affinity-purified with Raf-RBD-agarose and detected by immunoblotting for total Ras (Upper). Equal input was demonstrated by immunoblotting (Lower). (B) Bone marrow was harvested and starved as above, then stimulated with GM-CSF at concentrations of 0, 1, 3, and 10 ng/ml for 5 min and analyzed for the indicated proteins by immunoblotting.
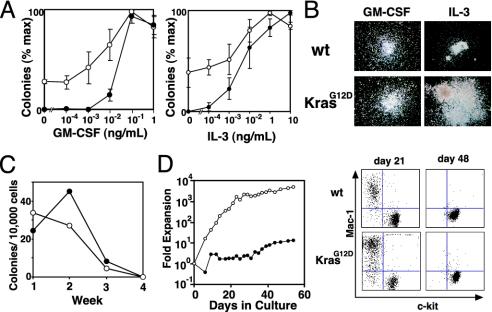
In vitro growth of Mx1-Cre KrasG12D (open circles) and control hematopoietic cells (filled circles). (A) Methylcellulose colony assays using GM-CSF or IL-3 at the indicated concentrations. Experiments were performed by using three mice of each genotype (±SEM). (B) Photomicrographs (original magnification ×40) of wild-type (wt) and mutant (KrasG12D) myeloid progenitor colonies grown in saturating concentrations of growth factors. (C) Colony formation with weekly serial replating of 10,000 unsorted bone marrow cells in methylcellulose. (D) Growth kinetics and flow cytometry of bone marrow cells cultured in the presence of IL-3.
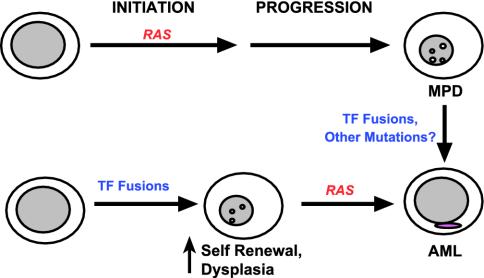
Proposed role of RAS mutations in MPD and AML. Our results and other human and murine data support the idea that initiating events that result in hyperactive Ras induce MPD (upper pathway). Transcription factor (TF) fusions are detected in some cases of CML at the time of evolution to blast crisis. Other acquired mutations are likely to exist as well. By contrast, leukemia-associated TF fusions may initiate AML by perturbing cell fates but appear to require a cooperating mutation that drives proliferation to cause overt disease. Here, oncogenic RAS (or activated FLT3) represent secondary mutations that contribute to leukemogenesis (lower pathway). The ability of oncogenic RAS to either initiate disease or cooperate with preexisting genetic lesions is likely to account for the finding of RAS mutations in myeloid malignancies with distinct phenotypes.
Similar articles
-
Conditional expression of oncogenic K-ras from its endogenous promoter induces a myeloproliferative disease.J Clin Invest. 2004 Feb;113(4):528-38. doi: 10.1172/JCI20476. J Clin Invest. 2004. PMID: 14966562 Free PMC article.
-
Hematopoiesis and leukemogenesis in mice expressing oncogenic NrasG12D from the endogenous locus.Blood. 2011 Feb 10;117(6):2022-32. doi: 10.1182/blood-2010-04-280750. Epub 2010 Dec 16. Blood. 2011. PMID: 21163920 Free PMC article.
-
Loss of wild-type Kras promotes activation of all Ras isoforms in oncogenic Kras-induced leukemogenesis.Leukemia. 2016 Jul;30(7):1542-51. doi: 10.1038/leu.2016.40. Epub 2016 Feb 29. Leukemia. 2016. PMID: 27055865 Free PMC article.
-
NOX2 inhibition reduces oxidative stress and prolongs survival in murine KRAS-induced myeloproliferative disease.Oncogene. 2019 Feb;38(9):1534-1543. doi: 10.1038/s41388-018-0528-1. Epub 2018 Oct 15. Oncogene. 2019. PMID: 30323311 Free PMC article.
-
Loss of Dnmt3a and endogenous Kras(G12D/+) cooperate to regulate hematopoietic stem and progenitor cell functions in leukemogenesis.Leukemia. 2015 Sep;29(9):1847-56. doi: 10.1038/leu.2015.85. Epub 2015 Mar 24. Leukemia. 2015. PMID: 25801914 Free PMC article.
Cited by
-
Cooperative loss of RAS feedback regulation drives myeloid leukemogenesis.Nat Genet. 2015 May;47(5):539-43. doi: 10.1038/ng.3251. Epub 2015 Mar 30. Nat Genet. 2015. PMID: 25822087 Free PMC article.
-
Constitutive MAP kinase activation in hematopoietic stem cells induces a myeloproliferative disorder.PLoS One. 2011;6(12):e28350. doi: 10.1371/journal.pone.0028350. Epub 2011 Dec 2. PLoS One. 2011. PMID: 22164275 Free PMC article.
-
Effect of Ras inhibition in hematopoiesis and BCR/ABL leukemogenesis.J Hematol Oncol. 2008 Jun 5;1:5. doi: 10.1186/1756-8722-1-5. J Hematol Oncol. 2008. PMID: 18577264 Free PMC article.
-
Cell-intrinsic depletion of Aml1-ETO-expressing pre-leukemic hematopoietic stem cells by K-Ras activating mutation.Haematologica. 2019 Nov;104(11):2215-2224. doi: 10.3324/haematol.2018.205351. Epub 2019 Apr 11. Haematologica. 2019. PMID: 30975913 Free PMC article.
-
Endogenous oncogenic Nras mutation initiates hematopoietic malignancies in a dose- and cell type-dependent manner.Blood. 2011 Jul 14;118(2):368-79. doi: 10.1182/blood-2010-12-326058. Epub 2011 May 17. Blood. 2011. PMID: 21586752 Free PMC article.
References
-
- Bos, J. L. (1989) Cancer Res. 49, 4682–4689. - PubMed
-
- Bourne, H. R., Sanders, D. A. & McCormick, F. (1990) Nature 348, 125–132. - PubMed
-
- Bourne, H. R., Sanders, D. A. & McCormick, F. (1991) Nature 349, 117–127. - PubMed
-
- Boguski, M. & McCormick, F. (1993) Nature 366, 643–653. - PubMed
-
- Serrano, M., Lin, A. W., McCurrach, M. E., Beach, D. & Lowe, S. W. (1997) Cell 88, 593–602. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
