

Anoxic fibroblasts activate a replication checkpoint that is bypassed by E1a
- PMID: 14645516
- PMCID: PMC309642
- DOI: 10.1128/MCB.23.24.9032-9045.2003
Anoxic fibroblasts activate a replication checkpoint that is bypassed by E1a
Abstract
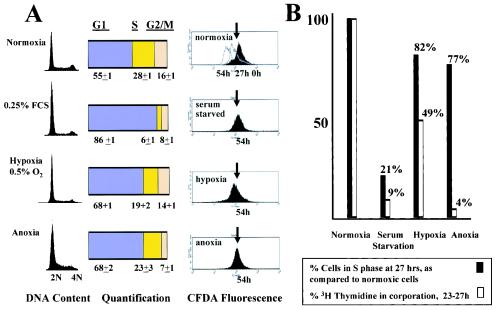
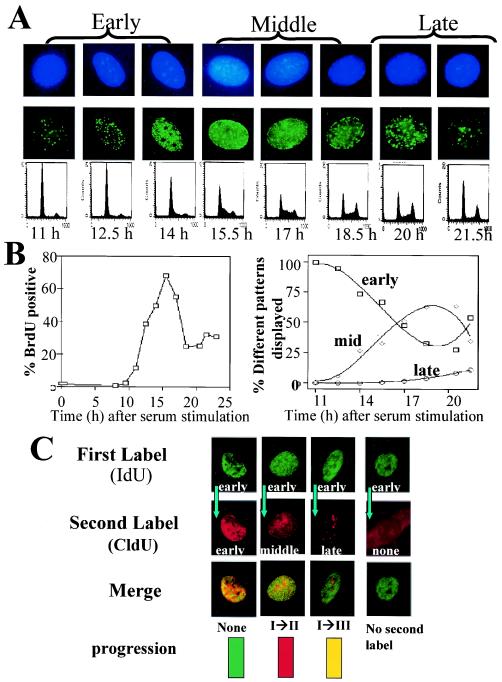
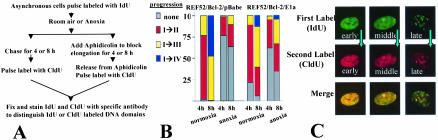
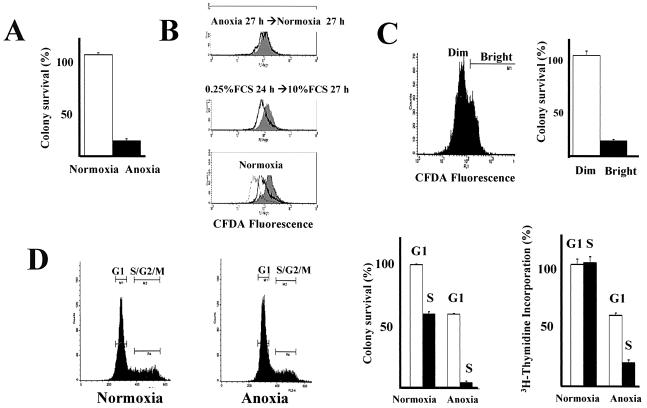
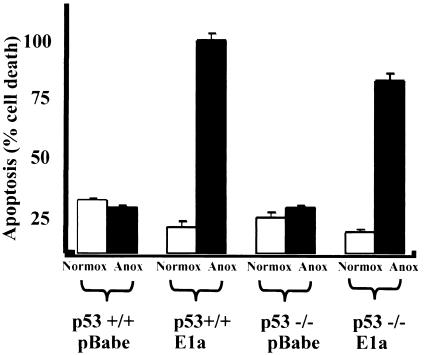
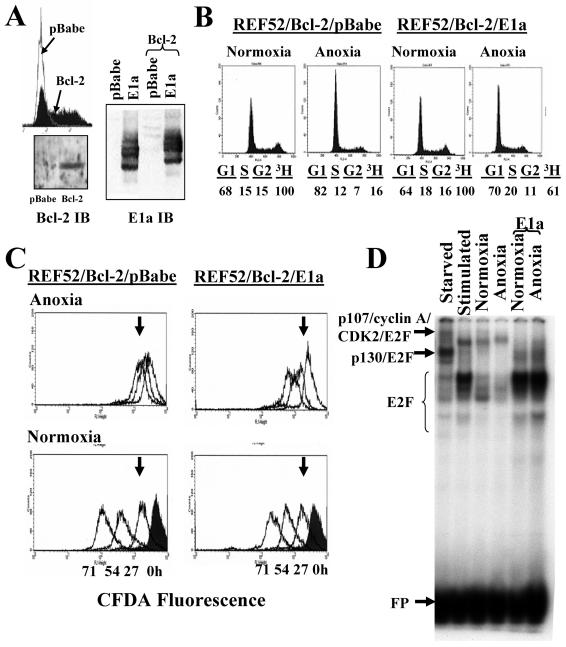
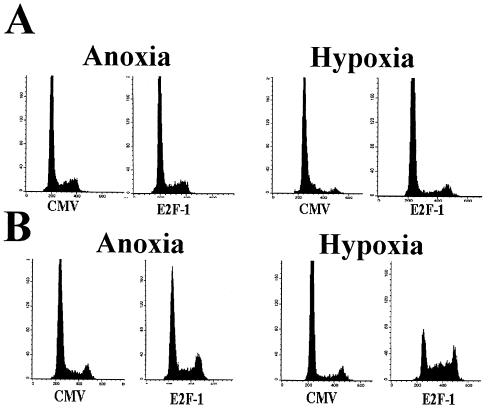
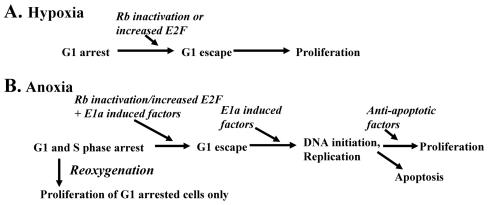
Little is known about cell cycle regulation in hypoxic cells, despite its significance. We utilized an experimentally tractable model to study the proliferative responses of rat fibroblasts when rendered hypoxic (0.5% oxygen) or anoxic (<0.01% oxygen). Hypoxic cells underwent G1 arrest, whereas anoxic cells also demonstrated S-phase arrest due to suppression of DNA initiation. Upon reoxygenation, only those cells arrested in G1 were able to resume proliferation. The oncoprotein E1a induced p53-independent apoptosis in anoxic cells, which when suppressed by Bcl-2 permitted proliferation despite anoxia. E1a expression led to marked increases in the transcription factor E2F, and overexpression of E2F-1 allowed proliferation in hypoxic cells, although it had minimal effect on the anoxic suppression of DNA initiation. We thus demonstrate two distinct cell cycle responses to low oxygen and suggest that alterations that lead to increased E2F can overcome hypoxic G1 arrest but that additional alterations, promoted by E1a expression, are necessary for neoplastic cells to proliferate despite anoxia.
Figures

Similar articles
-
[Changes in the activity of cyclin-kinase complexes governing cell transition from G1 phase to DNA replication phase in E1A + c-Ha-ras transformants transfected with the bcl-2 gene].Tsitologiia. 2003;45(2):149-57. Tsitologiia. 2003. PMID: 12722479 Russian.
-
[E1A oncogene effect on the ability of p21(Waf1) to regulate G1/S arrest in E1A-expressing transformants following irradiation].Tsitologiia. 2005;47(12):1063-70. Tsitologiia. 2005. PMID: 16706194 Russian.
-
Deregulation of p53/p21Cip1/Waf1 pathway contributes to polyploidy and apoptosis of E1A+cHa-ras transformed cells after gamma-irradiation.Oncogene. 1999 Oct 7;18(41):5611-9. doi: 10.1038/sj.onc.1202945. Oncogene. 1999. PMID: 10523840
-
Activated H-ras rescues E1A-induced apoptosis and cooperates with E1A to overcome p53-dependent growth arrest.Mol Cell Biol. 1995 Aug;15(8):4536-44. doi: 10.1128/MCB.15.8.4536. Mol Cell Biol. 1995. PMID: 7623844 Free PMC article.
-
Adenovirus and cell cycle control.Front Biosci. 2002 May 1;7:d1369-95. doi: 10.2741/ben. Front Biosci. 2002. PMID: 11991831 Review.
Cited by
-
ATM activation and signaling under hypoxic conditions.Mol Cell Biol. 2009 Jan;29(2):526-37. doi: 10.1128/MCB.01301-08. Epub 2008 Nov 3. Mol Cell Biol. 2009. PMID: 18981219 Free PMC article.
-
PP6C hotspot mutations in melanoma display sensitivity to Aurora kinase inhibition.Mol Cancer Res. 2014 Mar;12(3):433-9. doi: 10.1158/1541-7786.MCR-13-0422. Epub 2013 Dec 12. Mol Cancer Res. 2014. PMID: 24336958 Free PMC article.
-
Staphylococcus aureus induces hypoxia and cellular damage in porcine dermal explants.Infect Immun. 2015 Jun;83(6):2531-41. doi: 10.1128/IAI.03075-14. Epub 2015 Apr 6. Infect Immun. 2015. PMID: 25847960 Free PMC article.
-
Newborn hypoxia/anoxia inhibits cardiomyocyte proliferation and decreases cardiomyocyte endowment in the developing heart: role of endothelin-1.PLoS One. 2015 Feb 18;10(2):e0116600. doi: 10.1371/journal.pone.0116600. eCollection 2015. PLoS One. 2015. PMID: 25692855 Free PMC article.
-
Epithelial and mesenchymal tumor compartments exhibit in vivo complementary patterns of vascular perfusion and glucose metabolism.Neoplasia. 2007 Nov;9(11):900-8. doi: 10.1593/neo.07541. Neoplasia. 2007. PMID: 18030358 Free PMC article.
References
-
- Angus, S. P., L. J. Wheeler, S. A. Ranmal, X. Zhang, M. P. Markey, C. K. Mathews, and E. S. Knudsen. 2002. Retinoblastoma tumor suppressor targets dNTP metabolism to regulate DNA replication. J. Biol. Chem. 277:44376-44384. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous