

Functional characterization and cold sensitivity of T1313A, a new mutation of the skeletal muscle sodium channel causing paramyotonia congenita in humans
- PMID: 14617673
- PMCID: PMC1664790
- DOI: 10.1113/jphysiol.2003.053082
Functional characterization and cold sensitivity of T1313A, a new mutation of the skeletal muscle sodium channel causing paramyotonia congenita in humans
Abstract
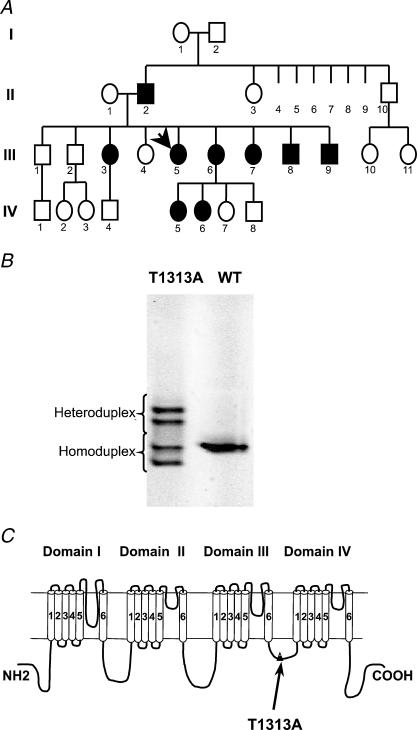
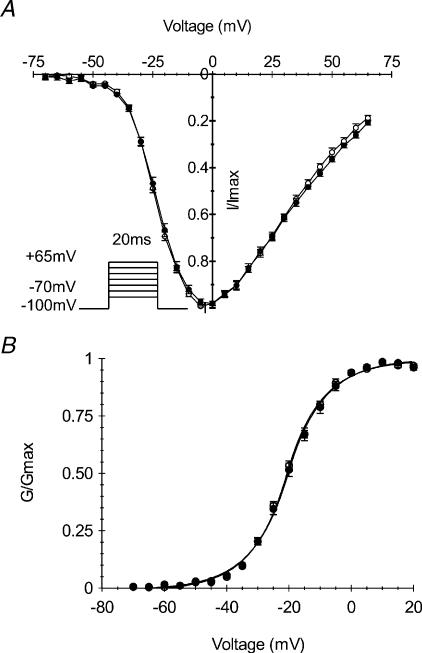
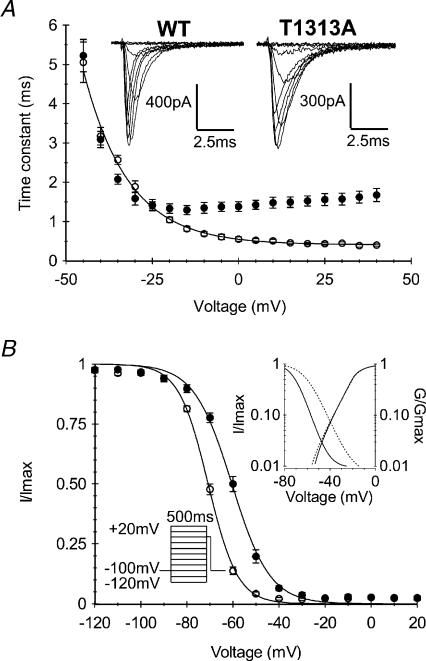
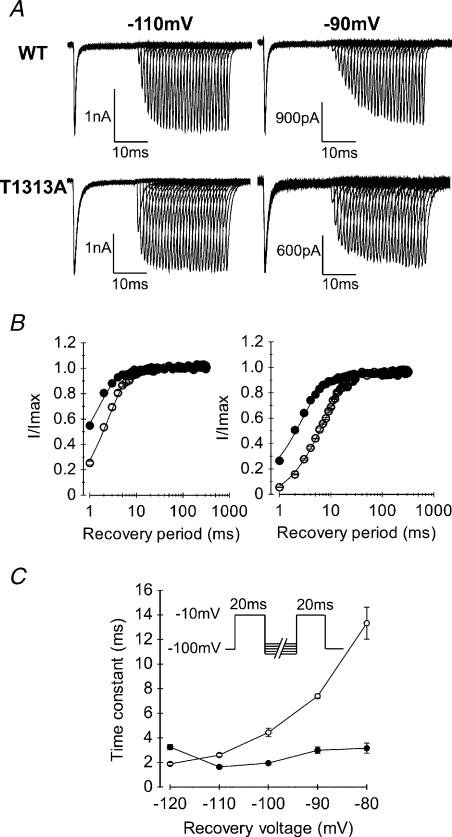
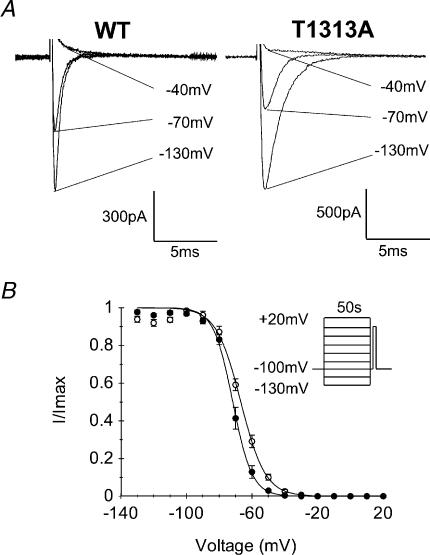
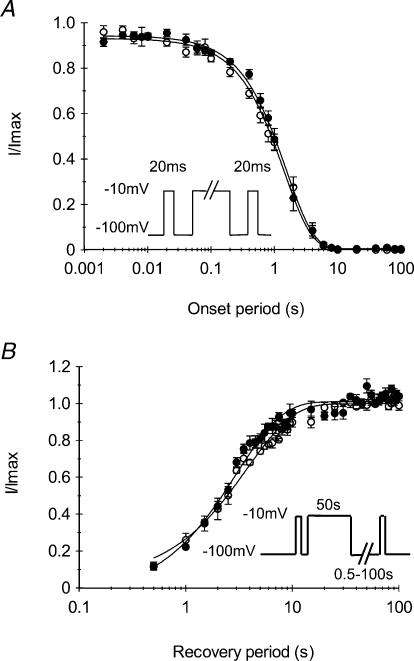
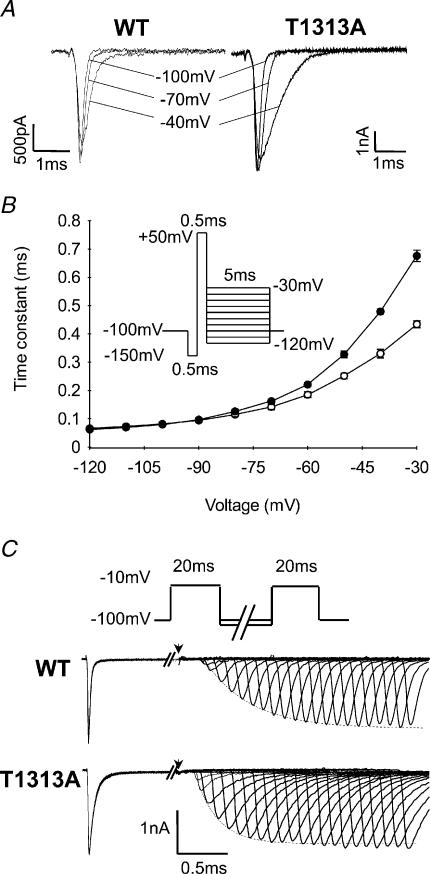
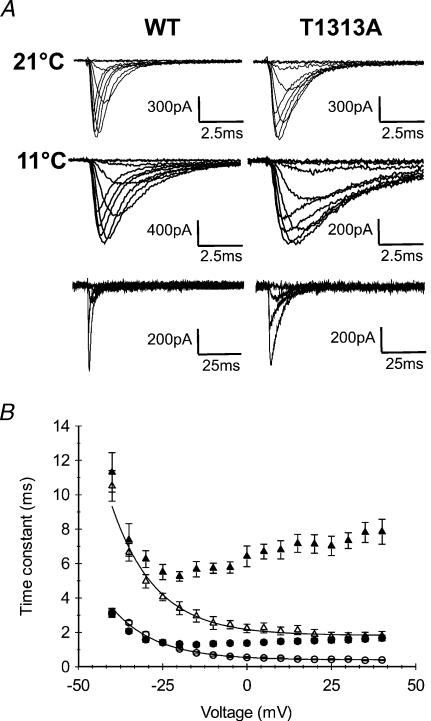
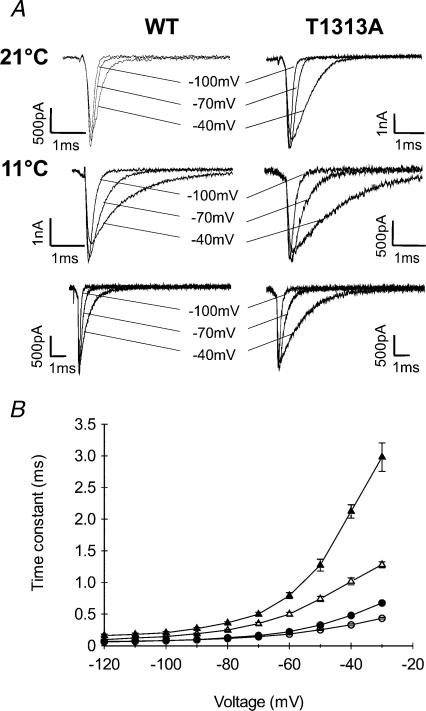
Paramyotonia congenita (PC) is a dominantly inherited skeletal muscle disorder caused by missense mutations in the SCN4A gene encoding the pore-forming alpha subunit (hSkM1) of the skeletal muscle Na+ channel. Muscle stiffness is the predominant clinical symptom. It is usually induced by exposure to cold and is aggravated by exercise. The most prevalent PC mutations occur at T1313 on DIII-DIV linker, and at R1448 on DIV-S4 of the alpha subunit. Only one substitution has been described at T1313 (T1313M), whereas four distinct amino-acid substitutions were found at R1448 (R1448C/H/P/S). We report herein a novel mutation at position 1313 (T1313A) associated with a typical phenotype of PC. We stably expressed T1313A or wild-type (hSkM1) channels in HEK293 cells, and performed a detailed study on mutant channel gating defects using the whole-cell configuration of the patch-clamp technique. T1313A mutation impaired Na+ channel fast inactivation: it slowed and reduced the voltage sensitivity of the kinetics, accelerated the recovery, and decreased the voltage-dependence of the steady state. Slow inactivation was slightly enhanced by the T1313A mutation: the voltage dependence was shifted toward hyperpolarization and its steepness was reduced compared to wild-type. Deactivation from the open state assessed by the tail current decay was only slowed at positive potentials. This may be an indirect consequence of disrupted fast inactivation. Deactivation from the inactivation state was hastened. The T1313A mutation did not modify the temperature sensitivity of the Na+ channel per se. However, gating kinetics of the mutant channels were further slowed with cooling, and reached levels that may represent the threshold for myotonia. In conclusion, our results confirm the role of T1313 residue in Na+ channel fast inactivation, and unveil subtle changes in other gating processes that may influence the clinical phenotype.
Figures
Similar articles
-
A1152D mutation of the Na+ channel causes paramyotonia congenita and emphasizes the role of DIII/S4-S5 linker in fast inactivation.J Physiol. 2005 Jun 1;565(Pt 2):415-27. doi: 10.1113/jphysiol.2004.081018. Epub 2005 Mar 24. J Physiol. 2005. PMID: 15790667 Free PMC article.
-
N1366S mutation of human skeletal muscle sodium channel causes paramyotonia congenita.J Physiol. 2017 Nov 15;595(22):6837-6850. doi: 10.1113/JP274877. Epub 2017 Oct 15. J Physiol. 2017. PMID: 28940424 Free PMC article.
-
Mechanisms of cold sensitivity of paramyotonia congenita mutation R1448H and overlap syndrome mutation M1360V.J Physiol. 2003 Mar 15;547(Pt 3):691-8. doi: 10.1113/jphysiol.2002.033928. Epub 2003 Jan 24. J Physiol. 2003. PMID: 12562902 Free PMC article.
-
Human skeletal muscle sodium channelopathies.Neurol Sci. 2005 Oct;26(4):194-202. doi: 10.1007/s10072-005-0461-x. Neurol Sci. 2005. PMID: 16193245 Review.
-
Paramyotonia Congenita with Persistent Distal and Facial Muscle Weakness: A Case Report with Literature Review.J Neuromuscul Dis. 2020;7(2):193-201. doi: 10.3233/JND-190440. J Neuromuscul Dis. 2020. PMID: 32083589 Review.
Cited by
-
Sodium channelopathies of skeletal muscle and brain.Physiol Rev. 2021 Oct 1;101(4):1633-1689. doi: 10.1152/physrev.00025.2020. Epub 2021 Mar 26. Physiol Rev. 2021. PMID: 33769100 Free PMC article. Review.
-
Cold and warmth intensify pain-linked sodium channel gating effects and persistent currents.J Gen Physiol. 2023 Sep 4;155(9):e202213312. doi: 10.1085/jgp.202213312. Epub 2023 Aug 2. J Gen Physiol. 2023. PMID: 37531097 Free PMC article.
-
Defective fast inactivation recovery of Nav 1.4 in congenital myasthenic syndrome.Ann Neurol. 2015 May;77(5):840-50. doi: 10.1002/ana.24389. Epub 2015 Mar 27. Ann Neurol. 2015. PMID: 25707578 Free PMC article.
-
Muscle channelopathies and critical points in functional and genetic studies.J Clin Invest. 2005 Aug;115(8):2000-9. doi: 10.1172/JCI25525. J Clin Invest. 2005. PMID: 16075040 Free PMC article. Review.
-
Changes in Resurgent Sodium Current Contribute to the Hyperexcitability of Muscles in Patients with Paramyotonia Congenita.Biomedicines. 2021 Jan 8;9(1):51. doi: 10.3390/biomedicines9010051. Biomedicines. 2021. PMID: 33430134 Free PMC article.
References
-
- Aldrich RW, Stevens CF. Inactivation of open and closed sodium channels determined separately. Cold Spring Harb Symp Quant Biol. 1983;48:147–153. - PubMed
-
- Barchi RL. Protein components of the purified sodium channel from rat skeletal muscle sarcolemma. J Neurochem. 1983;40:1377–1385. - PubMed
-
- Bendahhou S, Cummins TR, Kula RW, Fu YH, Ptacek LJ. Impairment of slow inactivation as a common mechanism for periodic paralysis in DIIS4-S5. Neurology. 2002;58:1266–1272. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases