

Prolonged kappa opioid receptor phosphorylation mediated by G-protein receptor kinase underlies sustained analgesic tolerance
- PMID: 14597630
- PMCID: PMC2131729
- DOI: 10.1074/jbc.M305796200
Prolonged kappa opioid receptor phosphorylation mediated by G-protein receptor kinase underlies sustained analgesic tolerance
Abstract
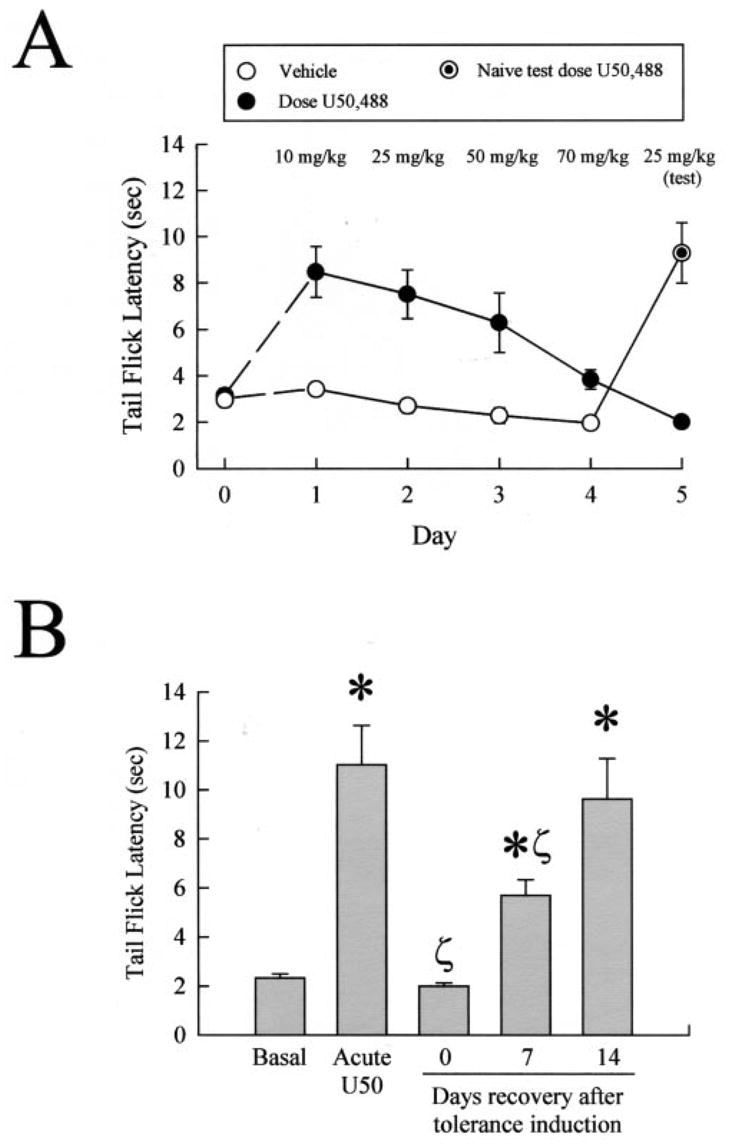
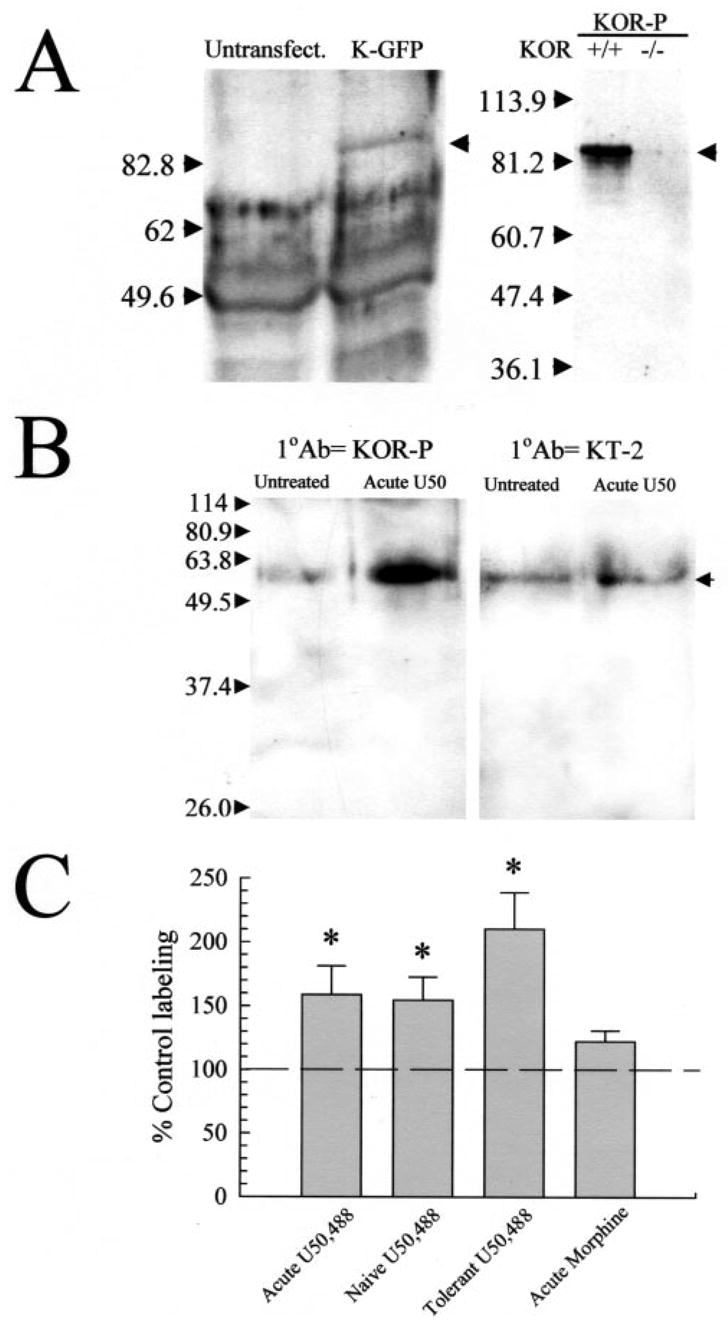
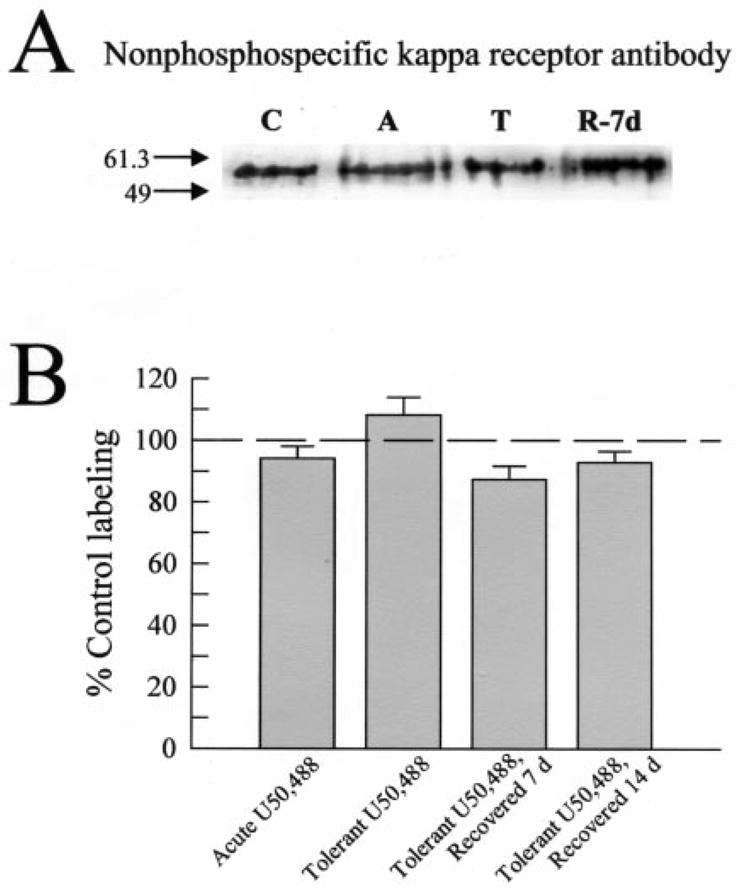
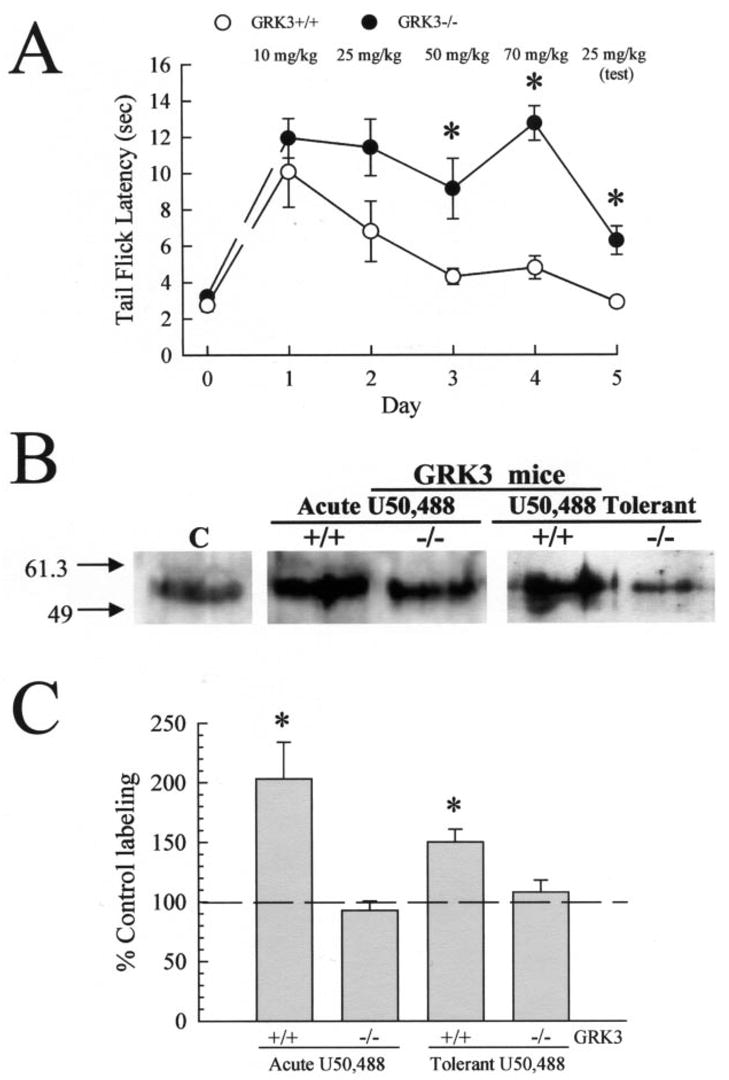
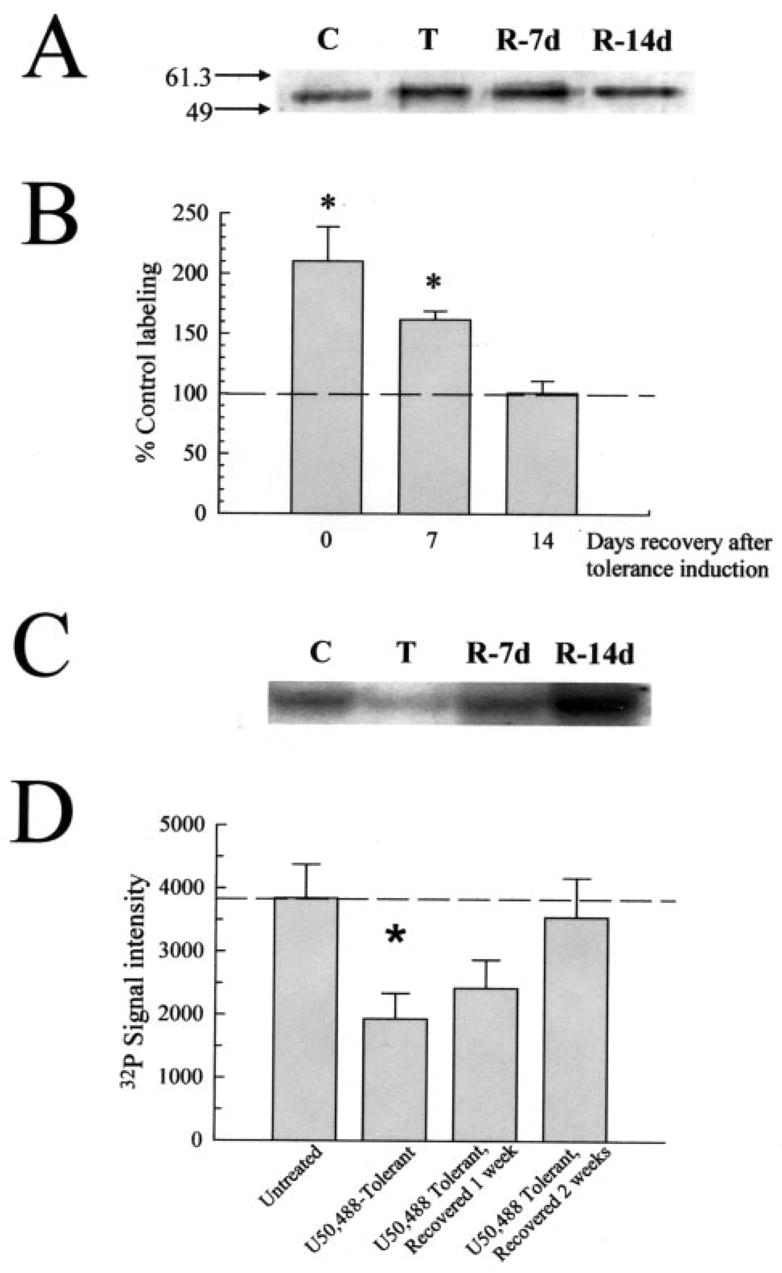
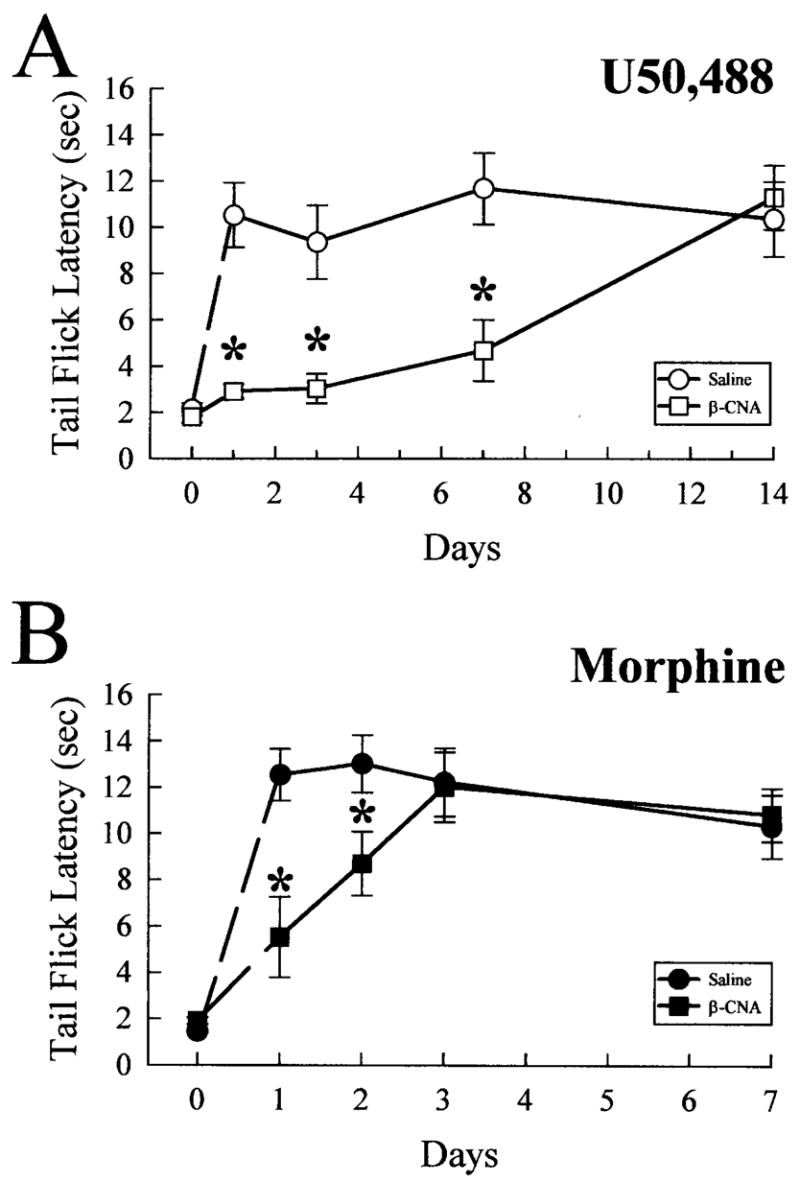
Kappa opioid receptor (KOR) desensitization was previously shown to follow agonist-dependent phosphorylation of serine 369 by G-protein receptor kinase (GRK) and beta-arrestin binding in transfected cells. To study the in vivo effects induced by phosphorylation of KOR(S369), C57Bl/6 mice were administered single or repeated doses of the KOR agonist, U50,488, and isolated brain glycoprotein was probed with an antibody, KOR-P, that specifically recognized phosphoserine 369 KOR. Western blot analysis using KOR-P antibody showed that labeling intensity increased after either single or repeated treatment of mice with U50,488 by 59 +/- 22% and 101 +/- 29%, respectively. In contrast, there was no change in labeling intensity by nonphosphoselective KOR antibodies following acute or chronic in vivo treatment with kappa agonist. Moreover, mice lacking GRK3 showed no increase in KOR-P labeling and developed significantly less analgesic tolerance following treatment with kappa agonist. The result suggests that tolerance to kappa agonists includes phosphorylation of serine 369 within KOR by GRK3. Recovery of analgesic potency and reduction of elevated KOR-P labeling in wild-type mice both required 2 weeks to return to base line. Consistent with these results, in vitro phosphorylation by GRK3 of KOR isolated from tolerant mice resulted in 46 +/- 7% less (32)P incorporation than in KOR isolated from untreated mice. In addition, in vitro (32)P incorporation returned to base line levels only in KOR isolated from tolerant mice allowed to recover for 2 weeks. The coincident reversal of analgesic tolerance and slow return to a basal phosphorylation state matched the regeneration rate of functional kappa receptors following irreversible antagonism and suggested that receptor replacement rather than dephosphorylation was required to restore sensitivity.
Figures
Similar articles
-
Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes.J Biol Chem. 2006 Jun 30;281(26):18081-9. doi: 10.1074/jbc.M513640200. Epub 2006 Apr 28. J Biol Chem. 2006. PMID: 16648139 Free PMC article.
-
Estrogen Regulation of GRK2 Inactivates Kappa Opioid Receptor Signaling Mediating Analgesia, But Not Aversion.J Neurosci. 2018 Sep 12;38(37):8031-8043. doi: 10.1523/JNEUROSCI.0653-18.2018. Epub 2018 Aug 3. J Neurosci. 2018. PMID: 30076211 Free PMC article.
-
Investigation of the role of βarrestin2 in kappa opioid receptor modulation in a mouse model of pruritus.Neuropharmacology. 2015 Dec;99:600-9. doi: 10.1016/j.neuropharm.2015.08.027. Epub 2015 Aug 25. Neuropharmacology. 2015. PMID: 26318102 Free PMC article.
-
Signaling underlying kappa opioid receptor-mediated behaviors in rodents.Front Neurosci. 2022 Nov 3;16:964724. doi: 10.3389/fnins.2022.964724. eCollection 2022. Front Neurosci. 2022. PMID: 36408401 Free PMC article. Review.
-
Antinociceptive Effects of Kappa-Opioid Receptor Agonists.Handb Exp Pharmacol. 2022;271:293-313. doi: 10.1007/164_2020_430. Handb Exp Pharmacol. 2022. PMID: 33387069 Review.
Cited by
-
Molecular Imaging of Opioid and Dopamine Systems: Insights Into the Pharmacogenetics of Opioid Use Disorders.Front Psychiatry. 2019 Sep 18;10:626. doi: 10.3389/fpsyt.2019.00626. eCollection 2019. Front Psychiatry. 2019. PMID: 31620026 Free PMC article. Review.
-
Opioid receptor interacting proteins and the control of opioid signaling.Curr Pharm Des. 2013;19(42):7333-47. doi: 10.2174/138161281942140105160625. Curr Pharm Des. 2013. PMID: 23448476 Free PMC article. Review.
-
Post-transcriptional regulation of opioid receptors in the nervous system.Front Biosci. 2004 May 1;9:1665-79. doi: 10.2741/1362. Front Biosci. 2004. PMID: 14977578 Free PMC article. Review.
-
Mu-opioid receptor desensitization: is morphine different?Br J Pharmacol. 2004 Nov;143(6):685-96. doi: 10.1038/sj.bjp.0705938. Epub 2004 Oct 25. Br J Pharmacol. 2004. PMID: 15504746 Free PMC article. Review.
-
Nicotine withdrawal and kappa-opioid receptors.Psychopharmacology (Berl). 2010 Jun;210(2):221-9. doi: 10.1007/s00213-009-1674-5. Epub 2009 Oct 6. Psychopharmacology (Berl). 2010. PMID: 19806344
References
-
- Gutstein HB, Akil H. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. Vol. 10. McGraw-Hill Co.; New York, NY: 2001. pp. 569–619.
-
- Nestler EJ. Curr Opin Neurobiol. 1997;7:713–719. - PubMed
-
- Law PY, Wong YH, Loh HH. Annu Rev Pharmacol Toxicol. 2000;40:389 –430. - PubMed
-
- Lefkowitz RJ. J Biol Chem. 1998;273:18677–18680. - PubMed
-
- Pei G, Kieffer BL, Lefkowitz RJ, Freedman NJ. Mol Pharmacol. 1995;48:173–177. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P01 DA015916-03/DA/NIDA NIH HHS/United States
- R01 DA011672-05/DA/NIDA NIH HHS/United States
- R03 DA 16656/DA/NIDA NIH HHS/United States
- P01 DA015916-010001/DA/NIDA NIH HHS/United States
- R01 DA 11672/DA/NIDA NIH HHS/United States
- T32 DA 07278/DA/NIDA NIH HHS/United States
- T32 DA007278/DA/NIDA NIH HHS/United States
- P01 DA015916-05/DA/NIDA NIH HHS/United States
- R01 DA011672-04/DA/NIDA NIH HHS/United States
- T32 DA007278-08/DA/NIDA NIH HHS/United States
- P01 DA015916-02/DA/NIDA NIH HHS/United States
- R01 DA011672-02/DA/NIDA NIH HHS/United States
- R01 DA011672-01A1/DA/NIDA NIH HHS/United States
- P01 DA015916/DA/NIDA NIH HHS/United States
- T32 DA007278-07/DA/NIDA NIH HHS/United States
- T32 DA007278-05/DA/NIDA NIH HHS/United States
- P01 DA015916-01/DA/NIDA NIH HHS/United States
- R01 DA 09040/DA/NIDA NIH HHS/United States
- P01 DA015916-04/DA/NIDA NIH HHS/United States
- R01 DA011672/DA/NIDA NIH HHS/United States
- R01 DA011672-03/DA/NIDA NIH HHS/United States
- T32 DA007278-06/DA/NIDA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
