

Identification of conserved regulatory elements by comparative genome analysis
- PMID: 12760745
- PMCID: PMC193685
- DOI: 10.1186/1475-4924-2-13
Identification of conserved regulatory elements by comparative genome analysis
Abstract
Background: For genes that have been successfully delineated within the human genome sequence, most regulatory sequences remain to be elucidated. The annotation and interpretation process requires additional data resources and significant improvements in computational methods for the detection of regulatory regions. One approach of growing popularity is based on the preferential conservation of functional sequences over the course of evolution by selective pressure, termed 'phylogenetic footprinting'. Mutations are more likely to be disruptive if they appear in functional sites, resulting in a measurable difference in evolution rates between functional and non-functional genomic segments.
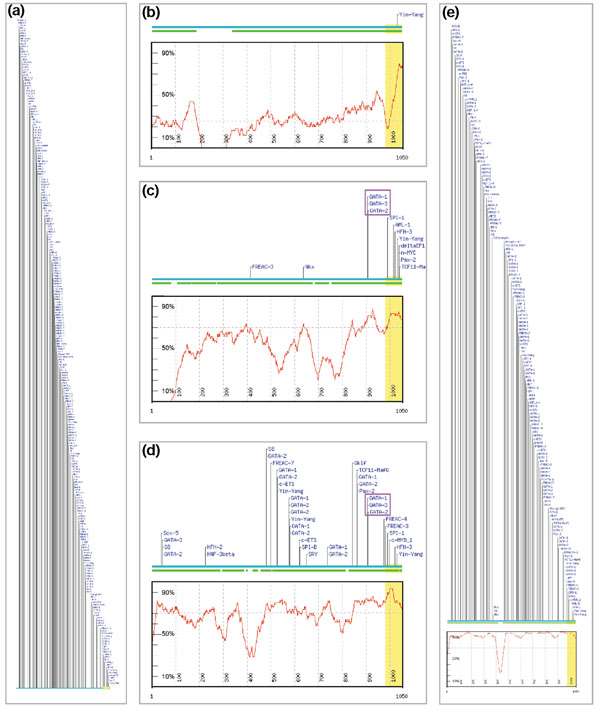
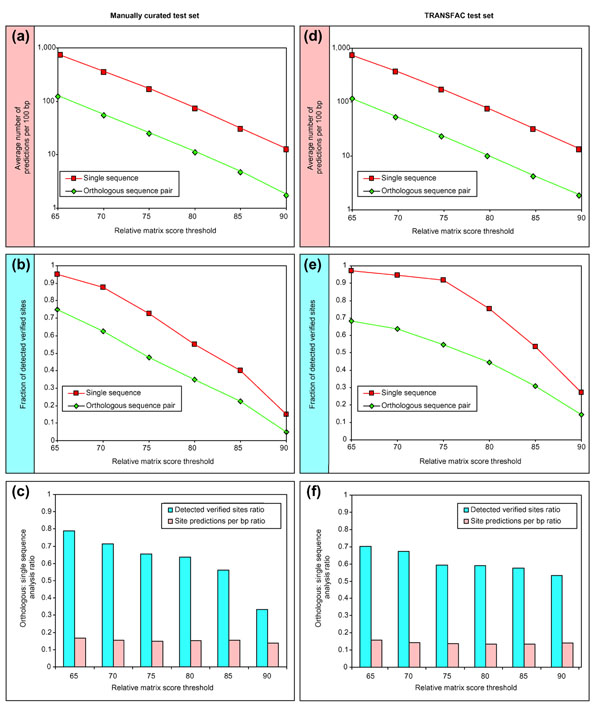
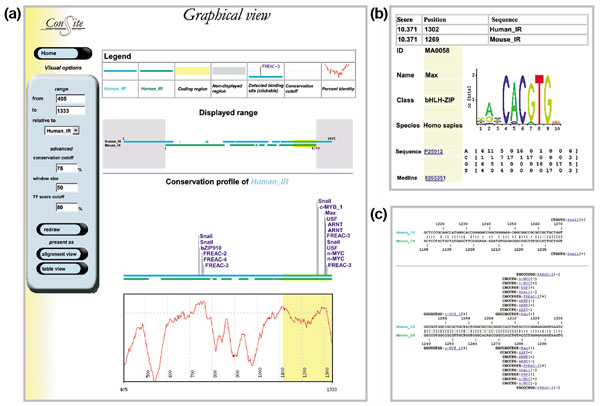
Results: We have devised a flexible suite of methods for the identification and visualization of conserved transcription-factor-binding sites. The system reports those putative transcription-factor-binding sites that are both situated in conserved regions and located as pairs of sites in equivalent positions in alignments between two orthologous sequences. An underlying collection of metazoan transcription-factor-binding profiles was assembled to facilitate the study. This approach results in a significant improvement in the detection of transcription-factor-binding sites because of an increased signal-to-noise ratio, as demonstrated with two sets of promoter sequences. The method is implemented as a graphical web application, ConSite, which is at the disposal of the scientific community at http://www.phylofoot.org/.
Conclusions: Phylogenetic footprinting dramatically improves the predictive selectivity of bioinformatic approaches to the analysis of promoter sequences. ConSite delivers unparalleled performance using a novel database of high-quality binding models for metazoan transcription factors. With a dynamic interface, this bioinformatics tool provides broad access to promoter analysis with phylogenetic footprinting.
Figures
Similar articles
-
CONREAL: conserved regulatory elements anchored alignment algorithm for identification of transcription factor binding sites by phylogenetic footprinting.Genome Res. 2004 Jan;14(1):170-8. doi: 10.1101/gr.1642804. Epub 2003 Dec 12. Genome Res. 2004. PMID: 14672977 Free PMC article.
-
Mulan: multiple-sequence local alignment and visualization for studying function and evolution.Genome Res. 2005 Jan;15(1):184-94. doi: 10.1101/gr.3007205. Epub 2004 Dec 8. Genome Res. 2005. PMID: 15590941 Free PMC article.
-
ConSite: web-based prediction of regulatory elements using cross-species comparison.Nucleic Acids Res. 2004 Jul 1;32(Web Server issue):W249-52. doi: 10.1093/nar/gkh372. Nucleic Acids Res. 2004. PMID: 15215389 Free PMC article.
-
The identification and functional characterisation of conserved regulatory elements in developmental genes.Brief Funct Genomic Proteomic. 2005 Feb;3(4):332-50. doi: 10.1093/bfgp/3.4.332. Brief Funct Genomic Proteomic. 2005. PMID: 15814024 Review.
-
Phylogenetic footprinting: a boost for microbial regulatory genomics.Protoplasma. 2012 Oct;249(4):901-7. doi: 10.1007/s00709-011-0351-9. Epub 2011 Nov 24. Protoplasma. 2012. PMID: 22113593 Review.
Cited by
-
A genome-wide cis-regulatory element discovery method based on promoter sequences and gene co-expression networks.BMC Genomics. 2013;14 Suppl 1(Suppl 1):S4. doi: 10.1186/1471-2164-14-S1-S4. Epub 2013 Jan 21. BMC Genomics. 2013. PMID: 23368633 Free PMC article.
-
Transposable elements donate lineage-specific regulatory sequences to host genomes.Cytogenet Genome Res. 2005;110(1-4):333-41. doi: 10.1159/000084965. Cytogenet Genome Res. 2005. PMID: 16093685 Free PMC article. Review.
-
Phylogenetic simulation of promoter evolution: estimation and modeling of binding site turnover events and assessment of their impact on alignment tools.Genome Biol. 2007;8(10):R225. doi: 10.1186/gb-2007-8-10-r225. Genome Biol. 2007. PMID: 17956628 Free PMC article.
-
Identifying transcriptional targets.Genome Biol. 2004;5(3):210. doi: 10.1186/gb-2004-5-3-210. Epub 2004 Feb 27. Genome Biol. 2004. PMID: 15005803 Free PMC article. Review.
-
Tracking evolution's footprints in the genome.J Biol. 2003;2(2):9. doi: 10.1186/1475-4924-2-9. Epub 2003 Jun 23. J Biol. 2003. PMID: 12818003 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
