

beta-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates beta-adrenoceptor switching from Gs to Gi
- PMID: 12552097
- PMCID: PMC298705
- DOI: 10.1073/pnas.262787199
beta-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates beta-adrenoceptor switching from Gs to Gi
Retraction in
-
Retraction for Baillie et al., β-Arrestin-mediated PDE4 cAMP phosphodiesterase recruitment regulates β-adrenoceptor switching from Gs to Gi.Proc Natl Acad Sci U S A. 2022 Apr 26;119(17):e2205198119. doi: 10.1073/pnas.2205198119. Epub 2022 Apr 26. Proc Natl Acad Sci U S A. 2022. PMID: 35471901 Free PMC article. No abstract available.
Abstract
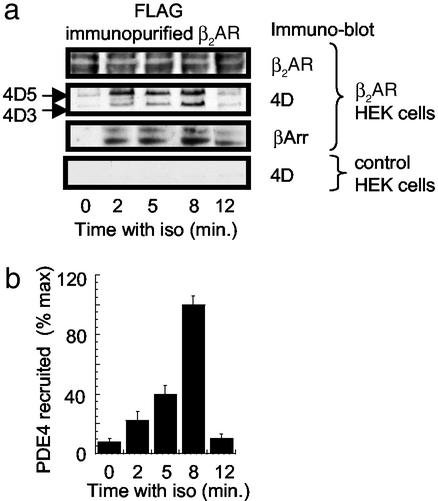
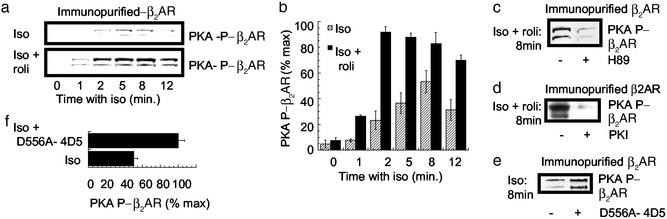
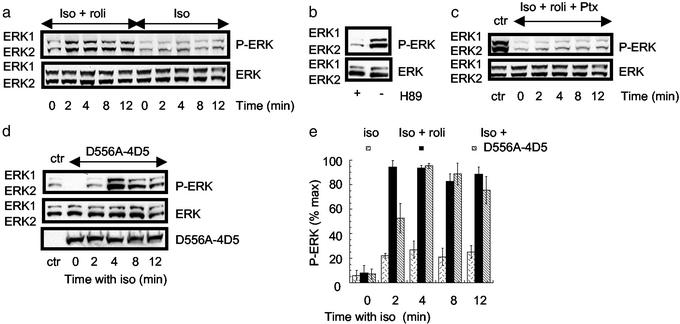
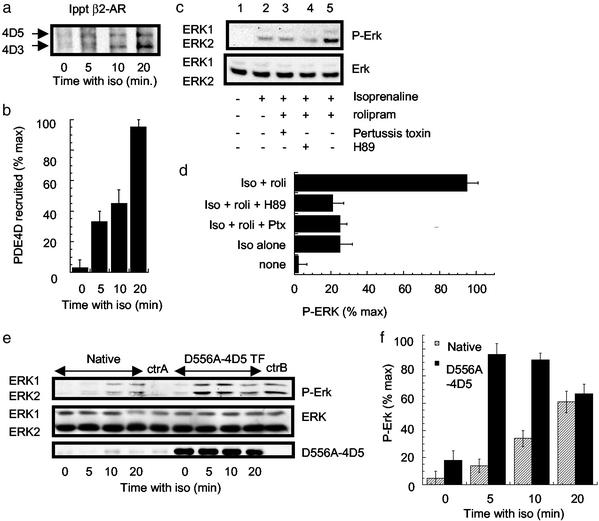
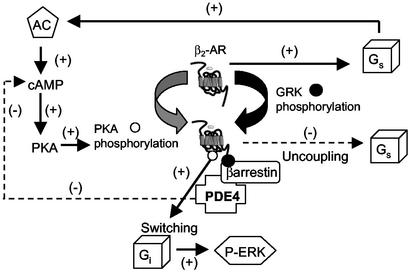
Phosphorylation of the beta(2) adrenoreceptor (beta(2)AR) by cAMP-activated protein kinase A (PKA) switches its predominant coupling from stimulatory guanine nucleotide regulatory protein (G(s)) to inhibitory guanine nucleotide regulatory protein (G(i)). beta-Arrestins recruit the cAMP-degrading PDE4 phosphodiesterases to the beta(2)AR, thus controlling PKA activity at the membrane. Here we investigate a role for PDE4 recruitment in regulating G protein switching by the beta(2)AR. In human embryonic kidney 293 cells overexpressing a recombinant beta(2)AR, stimulation with isoprenaline recruits beta-arrestins 1 and 2 as well as both PDE4D3 and PDE4D5 to the receptor and stimulates receptor phosphorylation by PKA. The PKA phosphorylation status of the beta(2)AR is enhanced markedly when cells are treated with the selective PDE4-inhibitor rolipram or when they are transfected with a catalytically inactive PDE4D mutant (PDE4D5-D556A) that competitively inhibits isoprenaline-stimulated recruitment of native PDE4 to the beta(2)AR. Rolipram and PDE4D5-D556A also enhance beta(2)AR-mediated activation of extracellular signal-regulated kinases ERK12. This is consistent with a switch in coupling of the receptor from G(s) to G(i), because the ERK12 activation is sensitive to both inhibitors of PKA (H89) and G(i) (pertussis toxin). In cardiac myocytes, the beta(2)AR also switches from G(s) to G(i) coupling. Treating primary cardiac myocytes with isoprenaline induces recruitment of PDE4D3 and PDE4D5 to membranes and activates ERK12. Rolipram robustly enhances this activation in a manner sensitive to both pertussis toxin and H89. Adenovirus-mediated expression of PDE4D5-D556A also potentiates ERK12 activation. Thus, receptor-stimulated beta-arrestin-mediated recruitment of PDE4 plays a central role in the regulation of G protein switching by the beta(2)AR in a physiological system, the cardiac myocyte.
Figures
Similar articles
-
RNA silencing identifies PDE4D5 as the functionally relevant cAMP phosphodiesterase interacting with beta arrestin to control the protein kinase A/AKAP79-mediated switching of the beta2-adrenergic receptor to activation of ERK in HEK293B2 cells.J Biol Chem. 2005 Sep 30;280(39):33178-89. doi: 10.1074/jbc.M414316200. Epub 2005 Jul 19. J Biol Chem. 2005. PMID: 16030021
-
Beta-arrestin-recruited phosphodiesterase-4 desensitizes the AKAP79/PKA-mediated switching of beta2-adrenoceptor signalling to activation of ERK.Biochem Soc Trans. 2005 Dec;33(Pt 6):1333-6. doi: 10.1042/BST0331333. Biochem Soc Trans. 2005. PMID: 16246112
-
Mapping binding sites for the PDE4D5 cAMP-specific phosphodiesterase to the N- and C-domains of beta-arrestin using spot-immobilized peptide arrays.Biochem J. 2007 May 15;404(1):71-80. doi: 10.1042/BJ20070005. Biochem J. 2007. PMID: 17288540 Free PMC article.
-
cAMP-specific phosphodiesterase-4D5 (PDE4D5) provides a paradigm for understanding the unique non-redundant roles that PDE4 isoforms play in shaping compartmentalized cAMP cell signalling.Biochem Soc Trans. 2007 Nov;35(Pt 5):938-41. doi: 10.1042/BST0350938. Biochem Soc Trans. 2007. PMID: 17956250 Review.
-
The role of ERK2 docking and phosphorylation of PDE4 cAMP phosphodiesterase isoforms in mediating cross-talk between the cAMP and ERK signalling pathways.Biochem Soc Trans. 2003 Dec;31(Pt 6):1186-90. doi: 10.1042/bst0311186. Biochem Soc Trans. 2003. PMID: 14641023 Review.
Cited by
-
β-Arrestin-2 desensitizes the transient receptor potential vanilloid 1 (TRPV1) channel.J Biol Chem. 2012 Oct 26;287(44):37552-63. doi: 10.1074/jbc.M112.391847. Epub 2012 Sep 5. J Biol Chem. 2012. PMID: 22952227 Free PMC article.
-
A biphasic and brain-region selective down-regulation of cyclic adenosine monophosphate concentrations supports object recognition in the rat.PLoS One. 2012;7(2):e32244. doi: 10.1371/journal.pone.0032244. Epub 2012 Feb 16. PLoS One. 2012. PMID: 22359674 Free PMC article.
-
Gravin orchestrates protein kinase A and β2-adrenergic receptor signaling critical for synaptic plasticity and memory.J Neurosci. 2012 Dec 12;32(50):18137-49. doi: 10.1523/JNEUROSCI.3612-12.2012. J Neurosci. 2012. PMID: 23238728 Free PMC article.
-
Molecular cloning and characterization of crustacean type-one dopamine receptors: D1alphaPan and D1betaPan.Comp Biochem Physiol B Biochem Mol Biol. 2006 Mar;143(3):294-301. doi: 10.1016/j.cbpb.2005.11.017. Epub 2006 Jan 19. Comp Biochem Physiol B Biochem Mol Biol. 2006. PMID: 16426885 Free PMC article.
-
Arthropod D2 receptors positively couple with cAMP through the Gi/o protein family.Comp Biochem Physiol B Biochem Mol Biol. 2007 Jan;146(1):9-19. doi: 10.1016/j.cbpb.2006.08.018. Epub 2006 Oct 10. Comp Biochem Physiol B Biochem Mol Biol. 2007. PMID: 17134931 Free PMC article.
References
-
- Rockman H A, Koch W J, Lefkowitz R J. Nature. 2002;415:206–212. - PubMed
-
- Daaka Y, Luttrell L M, Lefkowitz R J. Nature. 1997;390:88–91. - PubMed
-
- Lefkowitz R J, Pierce K L, Luttrell L M. Mol Pharmacol. 2002;62:971–974. - PubMed
-
- Zamah A M, Delahunty M, Luttrell L M, Lefkowitz R J. J Biol Chem. 2002;277:31249–31256. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
