

Microarray analysis of orthologous genes: conservation of the translational machinery across species at the sequence and expression level
- PMID: 12537549
- PMCID: PMC151285
- DOI: 10.1186/gb-2002-4-1-r4
Microarray analysis of orthologous genes: conservation of the translational machinery across species at the sequence and expression level
Abstract
Background: Genome projects have provided a vast amount of sequence information. Sequence comparison between species helps to establish functional catalogues within organisms and to study how they are maintained and modified across phylogenetic groups during evolution. Microarray studies allow us to determine groups of genes with similar temporal regulation and perhaps also common regulatory upstream regions for binding of transcription factors. The integration of sequence and expression data is expected to refine our current annotations and provide some insight into the evolution of gene regulation across organisms.
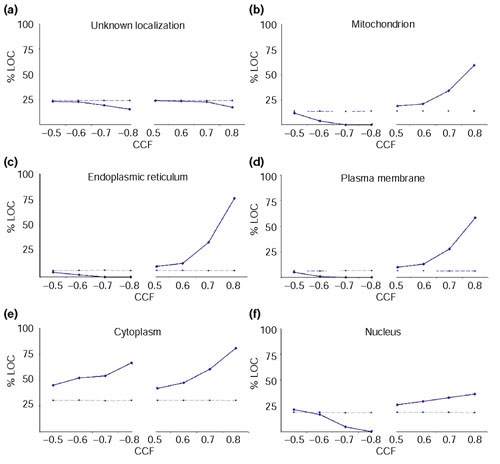
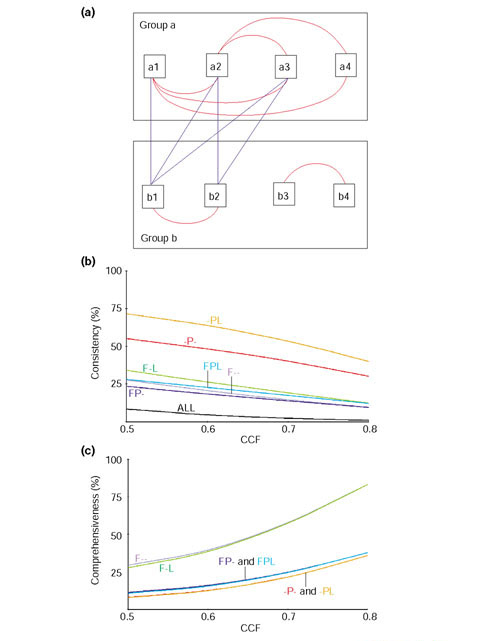
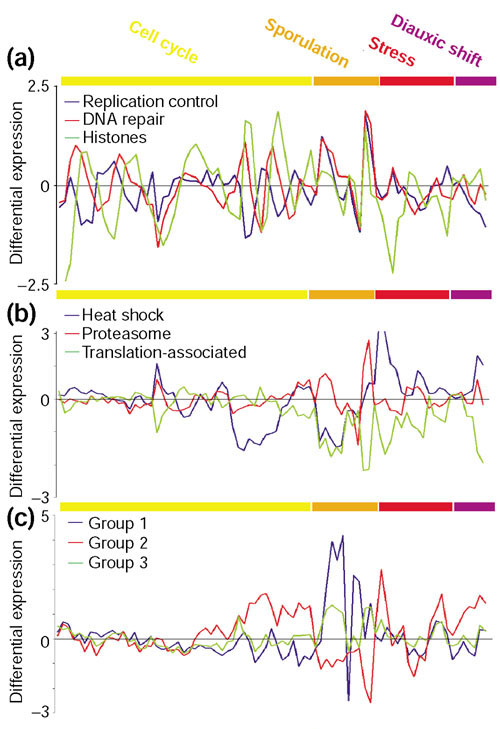
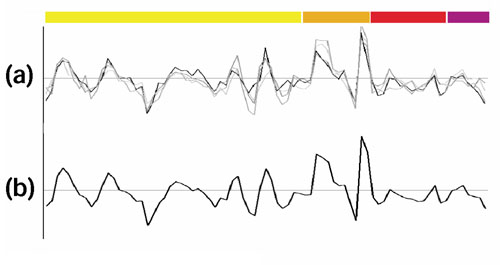
Results: We have investigated how well the protein subcellular localization and functional categories established from clustering of orthologous genes agree with gene-expression data in Saccharomyces cerevisiae. An increase in the resolution of biologically meaningful classes is observed upon the combination of experiments under different conditions. The functional categories deduced by sequence comparison approaches are, in general, preserved at the level of expression and can sometimes interact into larger co-regulated networks, such as the protein translation process. Differences and similarities in the expression between cytoplasmic-mitochondrial and interspecies translation machineries complement evolutionary information from sequence similarity.
Conclusions: Combination of several microarray experiments is a powerful tool for the identification of upstream regulatory motifs of yeast genes involved in protein synthesis. Comparison of these yeast co-regulated genes against the archaeal and bacterial operons indicates that the components of the protein translation process are conserved across organisms at the expression level with minor specific adaptations.
Figures

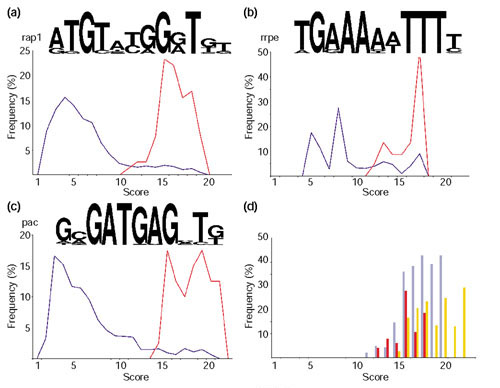
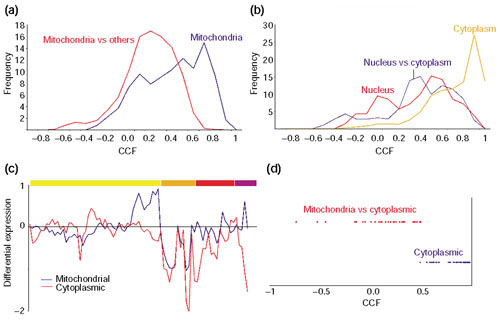
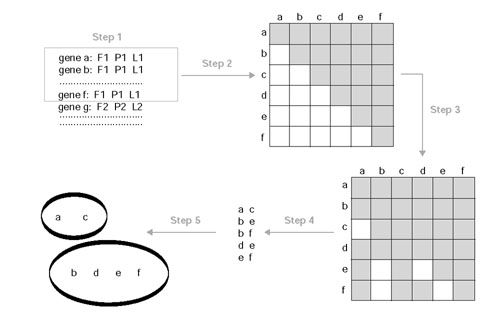
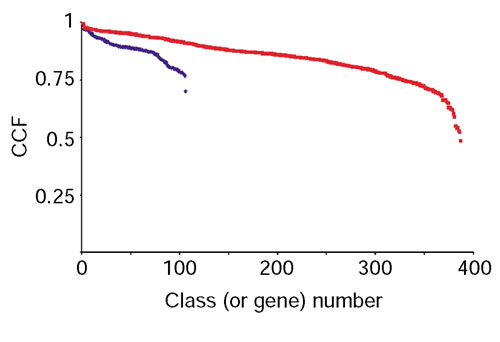
Similar articles
-
Expression studies and promoter analysis of the nuclear gene for mitochondrial transcription factor 1 (MTF1) in yeast.Curr Genet. 1999 Aug;36(1-2):37-48. doi: 10.1007/s002940050470. Curr Genet. 1999. PMID: 10447593
-
Sequencing and comparison of yeast species to identify genes and regulatory elements.Nature. 2003 May 15;423(6937):241-54. doi: 10.1038/nature01644. Nature. 2003. PMID: 12748633
-
Computational discovery of transcriptional regulatory modules in fungal ribosome biogenesis genes reveals novel sequence and function patterns.PLoS One. 2013;8(3):e59851. doi: 10.1371/journal.pone.0059851. Epub 2013 Mar 29. PLoS One. 2013. PMID: 23555806 Free PMC article.
-
A genomic perspective on protein families.Science. 1997 Oct 24;278(5338):631-7. doi: 10.1126/science.278.5338.631. Science. 1997. PMID: 9381173 Review.
-
Regulation of mitochondrial translation in yeast.Cell Mol Biol Lett. 2005;10(4):571-94. Cell Mol Biol Lett. 2005. PMID: 16341268 Review.
Cited by
-
DNA microarray data integration by ortholog gene analysis reveals potential molecular mechanisms of estrogen-dependent growth of human uterine fibroids.BMC Womens Health. 2007 Apr 2;7:5. doi: 10.1186/1472-6874-7-5. BMC Womens Health. 2007. PMID: 17407572 Free PMC article.
-
Genome adaptation to chemical stress: clues from comparative transcriptomics in Saccharomyces cerevisiae and Candida glabrata.Genome Biol. 2008;9(11):R164. doi: 10.1186/gb-2008-9-11-r164. Epub 2008 Nov 24. Genome Biol. 2008. PMID: 19025642 Free PMC article.
-
Cross-species hybridisation of human and bovine orthologous genes on high density cDNA microarrays.BMC Genomics. 2004 Oct 28;5:83. doi: 10.1186/1471-2164-5-83. BMC Genomics. 2004. PMID: 15511299 Free PMC article.
-
GO-2D: identifying 2-dimensional cellular-localized functional modules in Gene Ontology.BMC Genomics. 2007 Jan 24;8:30. doi: 10.1186/1471-2164-8-30. BMC Genomics. 2007. PMID: 17250772 Free PMC article.
-
High resolution RNA-seq profiling of genes encoding ribosomal proteins across different organs and developmental stages in Arabidopsis thaliana.Plant Direct. 2021 May 27;5(5):e00320. doi: 10.1002/pld3.320. eCollection 2021 May. Plant Direct. 2021. PMID: 34095740 Free PMC article.
References
-
- Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–474. - PubMed
-
- Klenk HP, Clayton RA, Tomb JF, White O, Nelson KE, Ketchum KA, Dodson RJ, Gwinn M, Hickey EK, Peterson JD, et al. The complete genome sequence of the hyperthermophilic, sulphate-reducing archaeon Archaeoglobus fulgidus. Nature. 1997;390:364–370. - PubMed
-
- Ruepp A, Graml W, Santos-Martinez ML, Koretke KK, Volker C, Mewes HW, Frishman D, Stocker S, Lupas AN, Baumeister W. The genome sequence of the thermoacidophilic scavenger Thermoplasma acidophilum. Nature. 2000;407:508–513. - PubMed
-
- The C. elegans Sequencing Consortium Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 1998;282:2012–2018. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
