

Cyclooxygenase-2 inhibition protects cultured cerebellar granule neurons from glutamate-mediated cell death
- PMID: 12042097
- PMCID: PMC1456322
- DOI: 10.1089/089771502753754091
Cyclooxygenase-2 inhibition protects cultured cerebellar granule neurons from glutamate-mediated cell death
Abstract
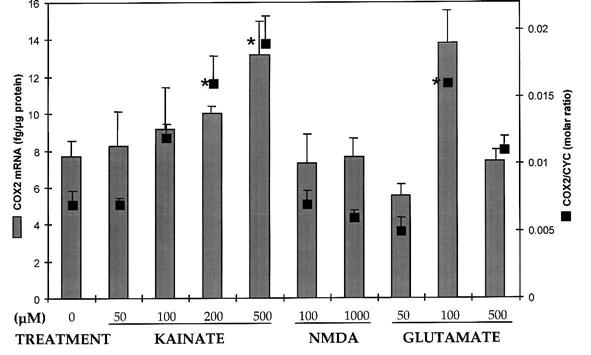
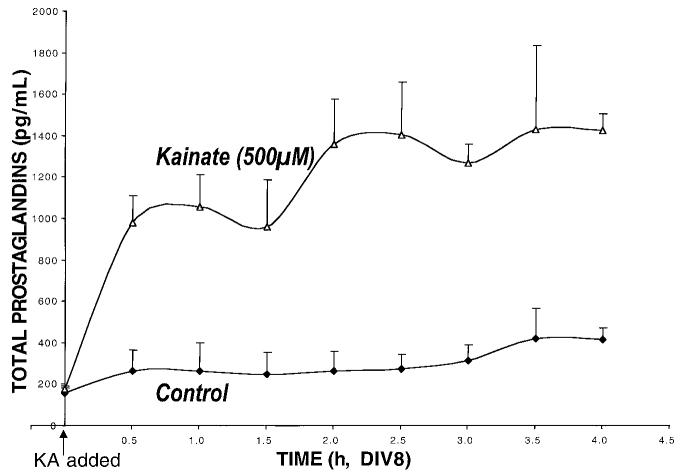
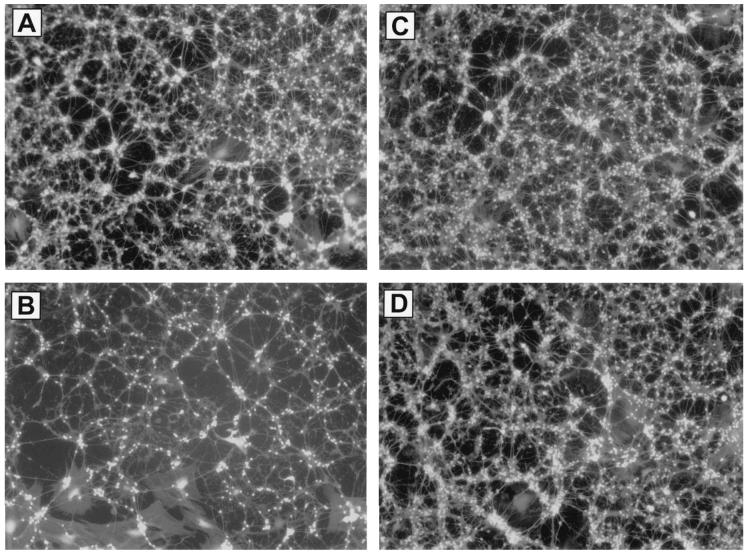
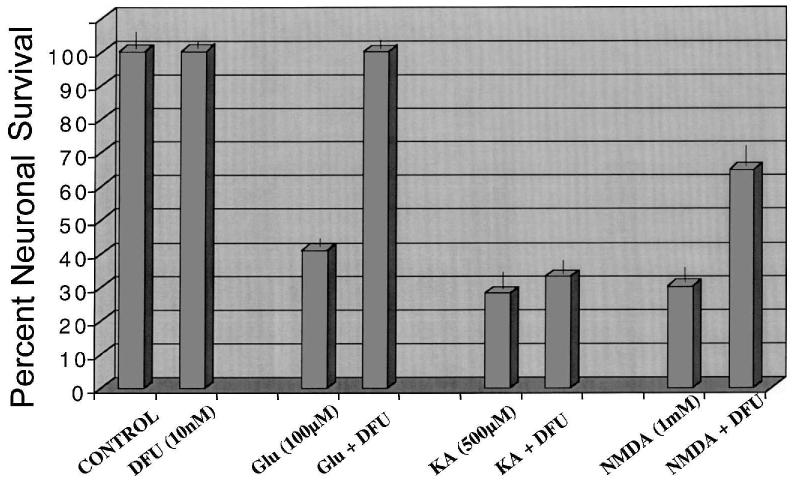
Primary insults to the brain can initiate glutamate release that may result in excitotoxicity followed by neuronal cell death. This secondary process is mediated by both N-methyl-D-aspartate (NMDA) and non-NMDA receptors in vivo and requires new gene expression. Neuronal cyclooxygenase-2 (COX2) expression is upregulated following brain insults, via glutamatergic and inflammatory mechanisms. The products of COX2 are bioactive prostanoids and reactive oxygen species that may play a role in neuronal survival. This study explores the role of neuronal COX2 in glutamate excitotoxicity using cultured cerebellar granule neurons (day 8 in vitro). Treatment with excitotoxic concentrations of glutamate or kainate transiently induced COX2 mRNA (two- and threefold at 6 h, respectively, p < 0.05, Dunnett) and prostaglandin production (five- and sixfold at 30 min, respectively, p < 0.05, Dunnett). COX2 induction peaked at toxic concentrations of these excitatory amino acids. Surprisingly, NMDA, L-quisqualate, and trans-ACPD did not induce COX2 mRNA at any concentration tested. The glutamate receptor antagonist NBQX (5 microM, AMPA/kainate receptor) completely inhibited kainate-induced COX2 mRNA and partially inhibited glutamate-induced COX2 (p < 0.05, Dunnett). Other glutamate receptor antagonists, such as MK-801 (1 microM, NMDA receptor) or MCPG (500 microM, class 1 metabotropic receptors), partially attenuated glutamate-induced COX2 mRNA. These antagonists all reduced steady-state COX2 mRNA (p < 0.05, Dunnett). To determine whether COX2 might be an effector of excitotoxic cell death, cerebellar granule cells were pretreated (24 h) with the COX2-specific enzyme inhibitor, DFU (5,5-dimethyl-3-(3-fluorophenyl)-4-(4-methylsulphonyl) phenyl-2((5)H)-furanone) prior to glutamate challenge. DFU (1 to 1000 nM) completely protected cultured neurons from glutamate-mediated neurotoxicity. Approximately 50% protection from NMDA-mediated neurotoxicity, and no protection from kainate-mediated neurotoxicity was observed. Therefore, glutamate-mediated COX2 induction contributes to excitotoxic neuronal death. These results suggest that glutamate, NMDA, and kainate neurotoxicity involve distinct excitotoxic pathways, and that the glutamate and NMDA pathways may intersect at the level of COX2.
Figures


Similar articles
-
Cyclooxygenase-2 contributes to N-methyl-D-aspartate-mediated neuronal cell death in primary cortical cell culture.J Pharmacol Exp Ther. 2000 May;293(2):417-25. J Pharmacol Exp Ther. 2000. PMID: 10773011
-
5-Lipoxygenase and cyclooxygenase mRNA expression in rat hippocampus:early response to glutamate receptor activation by kainate.Exp Gerontol. 2000 Dec;35(9-10):1201-9. doi: 10.1016/s0531-5565(00)00152-2. Exp Gerontol. 2000. PMID: 11113602
-
Evidence that the early loss of membrane protein kinase C is a necessary step in the excitatory amino acid-induced death of primary cortical neurons.J Neurochem. 1997 Apr;68(4):1400-12. doi: 10.1046/j.1471-4159.1997.68041400.x. J Neurochem. 1997. PMID: 9084410
-
[Protective effects of N-methyl-D-aspartate against neuronal damages].Nihon Shinkei Seishin Yakurigaku Zasshi. 2002 Oct;22(5):153-8. Nihon Shinkei Seishin Yakurigaku Zasshi. 2002. PMID: 12451685 Review. Japanese.
-
Molecular mechanisms of glutamate receptor-mediated excitotoxic neuronal cell death.Mol Neurobiol. 2001 Aug-Dec;24(1-3):107-29. doi: 10.1385/MN:24:1-3:107. Mol Neurobiol. 2001. PMID: 11831548 Review.
Cited by
-
Investigational and Experimental Drugs to Treat Obsessive-Compulsive Disorder.J Exp Pharmacol. 2021 Jan 5;12:695-706. doi: 10.2147/JEP.S255375. eCollection 2020. J Exp Pharmacol. 2021. PMID: 33447096 Free PMC article. Review.
-
NSAIDs in the treatment and/or prevention of neurological disorders.Inflammopharmacology. 2012 Jun;20(3):159-67. doi: 10.1007/s10787-011-0116-2. Epub 2012 Jan 10. Inflammopharmacology. 2012. PMID: 22231719 Review.
-
The evolution of the pilocarpine animal model of status epilepticus.Heliyon. 2020 Jul 28;6(7):e04557. doi: 10.1016/j.heliyon.2020.e04557. eCollection 2020 Jul. Heliyon. 2020. PMID: 32775726 Free PMC article. Review.
-
A liquid chromatography/mass spectrometric method for simultaneous analysis of arachidonic acid and its endogenous eicosanoid metabolites prostaglandins, dihydroxyeicosatrienoic acids, hydroxyeicosatetraenoic acids, and epoxyeicosatrienoic acids in rat brain tissue.J Pharm Biomed Anal. 2007 Feb 19;43(3):1122-34. doi: 10.1016/j.jpba.2006.10.009. Epub 2006 Nov 27. J Pharm Biomed Anal. 2007. PMID: 17125954 Free PMC article.
-
Macrophage migration inhibitory factor facilitates prostaglandin E2 production of astrocytes to tune inflammatory milieu following spinal cord injury.J Neuroinflammation. 2019 Apr 13;16(1):85. doi: 10.1186/s12974-019-1468-6. J Neuroinflammation. 2019. PMID: 30981278 Free PMC article.
References
-
- ADAMS J, COLLACO-MORAES Y, DEBELLEROCHE JS. Cyclooxygenase-2 induction in cerebral cortex: an intracellular response to synaptic excitation. J. Neurochem. 1996;66:6–13. - PubMed
-
- BAGETTA G, CORASANITI MT, PAOLETTI AM, et al. HIV-1 gp120-induced apoptosis in the rat neocortex involves enhanced expression of cyclooxygenase type 2 (COX-2) Biochem Biophys. Res. Commun. 1998;244:819–824. - PubMed
-
- BAIK EJ, KIM EJ, LEE SH, et al. Cyclooxygenase-2 selective inhibitors aggravate kainic acid induced seizure and neuronal cell death in the hippocampus. Brain Res. 1999;843:118–129. - PubMed
-
- BANAUDHA K, MARINI AM. AMPA prevents glutamate-induced neurotoxicity and apoptosis in cultured cerebellar granule cell neurons. Neurotoxicity Res. 2000;2:51–61. - PubMed
-
- BEAMAN-HALL CM, LEAHY JC, BENMANSOUR S, et al. Glia modulate NMDA-mediated signaling in primary cultures of cerebellar granule cells. J. Neurochem. 1998;71:1993–2005. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
